Desymmetrizations of meso-tert-norbornenols by rhodium(I)-catalyzed enantioselective retro-allylations†‡
Michael
Waibel
and
Nicolai
Cramer
*
Laboratory of Organic Chemistry, ETH Zurich, HCI H 304, Wolfgang-Pauli-Strasse 10, 8093 Zurich, Switzerland. E-mail: nicolai.cramer@org.chem.ethz.ch; Fax: +41 44 632 1328; Tel: +41 44 632 6412
First published on 31st August 2010
Abstract
Rhodium-catalyzed desymmetrizations of meso-tert-norbornenols by retro-allylations generate allyl-rhodium species that allow for a rich and diverse downstream reactivity.
The allylation of carbonyl groups is a reaction of fundamental importance to organic chemistry.1 On the other hand, the reverse reaction pathway—retro-allylation from tertiary homoallylic alcohols—is a process observed with a range of main group and transition-metals.2 Yorimitsu and Oshima disclosed in seminal contributions that the released late transition-metal allyl species can be used in coupled allylation processes.3 Contrasting carbonyl allylation reactions,4catalytic enantioselective variants of retro-allylations are limited to two examples. Hayashi and co-workers demonstrated kinetic resolutions of racemic tertiary alcohols with a chiral rhodium catalyst.5 We have recently reported one example of an enantioselective palladium(0)-catalyzed arylative retro-allylation allowing the preparation of a highly substituted cyclohexene using a taddol-based phosphoramidite ligand.6
Herein, we report the complementary reaction profile of allyl rhodium species 3s/3p generated by desymmetrization of bicyclic meso-tert-norbornenols 1 and illustrate their synthetic potential and variable reactivity (Scheme 1).
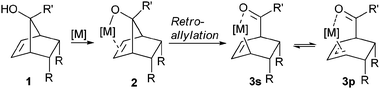 | ||
| Scheme 1 Desymmetrization of meso-tert-norbornenols 1 by metal promoted retro-allylation. | ||
A simultaneous coordination of the alkoxide and the olefin moiety7 of 4 to a chiral Rh(I)-complex induces an enantioselective retro-allylative C–C bond cleavage leading to allyl rhodium species either as their σ- (5s) or a π-bound (5p) complex (Scheme 2).8β-Hydride elimination forms regioisomeric dienes 69 and a rhodium hydride species, which in turn, is prone to re-addition to the formed diene.10 Re-additions in a 1,2- or 1,4-fashion could lead to enolate 7 and subsequent metal or base promoted isomerization yields more stable enone 8 as the terminal product. In view of the facile reversibility and the small differences in activation barriers of such additions/eliminations of rhodium hydrides to 6, we sought to explore conditions to address the selective formation of specific product branches.
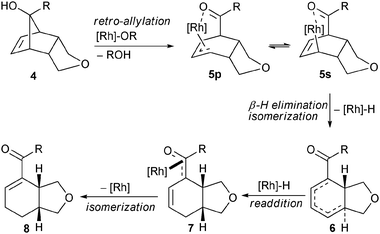 | ||
| Scheme 2 Proposed pathway for the formation of enone 8. | ||
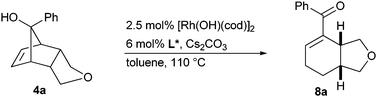
Refluxing a solution of 4a in the presence of 2.5 mol% [Rh(cod)(OH)]2, 6 mol% (R)-Binap and caesium carbonate11 in toluene gave ring opened enone 8a in 79% yield, albeit in a moderate enantiomeric ratio of 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶25 (Table 1, entry 1). Screening of a range of chiral diphosphines (Scheme 3) led to the identification of Josiphos L6 as a promising ligand giving 8a with an er of 91∶9 (entry 9). Performing the reaction at 115 °C and replacing toluene with chlorobenzene increased the yield from initially 36% to 74% (entry 10).
∶25 (Table 1, entry 1). Screening of a range of chiral diphosphines (Scheme 3) led to the identification of Josiphos L6 as a promising ligand giving 8a with an er of 91∶9 (entry 9). Performing the reaction at 115 °C and replacing toluene with chlorobenzene increased the yield from initially 36% to 74% (entry 10).
| Entry | Ligand L* | % Yieldb | er c |
|---|---|---|---|
| a Conditions: 0.05 mmol 4a, 2.5 mol% [Rh(OH)(cod)]2, 6.0 mol% L*, 1.0 equiv. Cs2CO3, toluene (0.15 M), 110 °C, 12 h. b Isolated product 8a. c By HPLC with a CSP (sign of the optical rotation). d At 120 °C. e PhCl instead of toluene. f At 115 °C. | |||
| 1 | (R)-Binap | 79 | 75∶25 (−) |
| 2 | (R)-H8-Binap (L1) | 83 | 78∶22 (−) |
| 3 | (R)-Segphos (L2) | 89 | 76∶24 (−) |
| 6d | (R)-DTBM-Segphos (L3) | 37 | 93∶7 (−) |
| 7 | (R)-DM-Segphos (L4) | 83 | 82∶18 (−) |
| 8 | (R)-Difluorphos (L5) | 83 | 79∶21 (−) |
| 9 | L6 | 36 | 91∶9 (+) |
| 10e,f | L6 | 74 | 91∶9 (+) |
| 11e,f | ent-L7 | 62 | 61∶39 (−) |
| 12e,f | ent-L8 | 71 | 90∶10 (−) |
| 13e,f | ent-L9 | 65 | 74∶26 (−) |
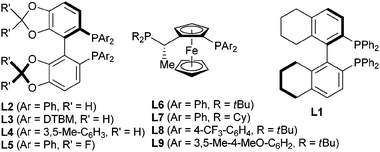 | ||
| Scheme 3 Utilized ligands L* (DTBM = 3,5-tBu-4-MeO-C6H2). | ||
With the optimized conditions, we then explored the influence of substituent R on the tertiary alcohol (Table 2). Aromatic groups with different steric and electronic properties are well tolerated and have little impact on the reactivity and selectivity of the process (entries 1–5). Although substituent R is oriented away from the reaction site, small alkyl groups cause diminished enantioselectivities (entries 6 and 7).
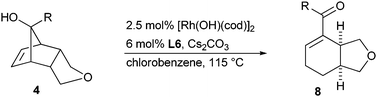
| Entry | 4 | R | 8 | % Yieldb | er c |
|---|---|---|---|---|---|
| a Conditions: 0.05 mmol 4, 2.5 mol% [Rh(OH)(cod)]2, 6.0 mol% L6, 1.0 equiv. Cs2CO3, PhCl (0.15 M), 115 °C, 12 h. b Isolated product 8. c By HPLC with a CSP (sign of the optical rotation). d ent-L6 was used. | |||||
| 1 | 4a | Ph | 8a | 74 | 91∶9 (+) |
| 2d | 4b | 4-MeO–Ph | ent-8b | 65 | 91∶9 (−) |
| 3 | 4c | 4-F–Ph | 8c | 84 | 91∶9 (+) |
| 4 | 4d | 2-Me–Ph | 8d | 80 | 88∶12 (+) |
| 5 | 4e | 1-Naphthyl | 8e | 88 | 90∶10 (+) |
| 6 | 4f | Me | 8f | 85 | 80∶20 (+) |
| 7 | 4g | Bu | 8g | 86 | 85∶15 (+) |
Noteworthy, when the base was omitted in the retro-allylation reaction of 4a, regio-isomeric β-hydride elimination of 5 became the dominant pathway and led to presumed diene 9 (Scheme 4). Subsequent 1,4-addition might form allyl-rhodium species 10, which again undergoes H elimination accounting for the observed diene 11, formed in 77% yield and an er of 78∶22. On the other hand, when vinyl substituted tert-alcohol 4h was submitted to the reaction conditions, formation of diene 13 as well as aromatic product 14 was observed (Scheme 5). This suggests a preferential re-addition of the rhodium hydride species to the terminal, activated olefin instead of the cyclohexadiene moiety of 6h.12 Enolate 12 might subsequently isomerize to the more stable enone 13 or aromatize to arene 14. Fine-tuning of the reaction condition by the addition of ten equivalents of cyclohexene mitigated the undesired oxidation to arene 14.
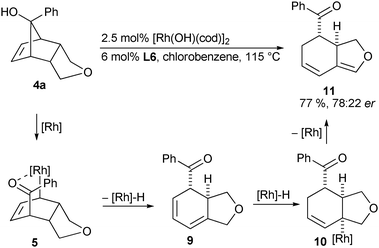 | ||
| Scheme 4 Proposed reaction pathways for the formation of diene 11. | ||
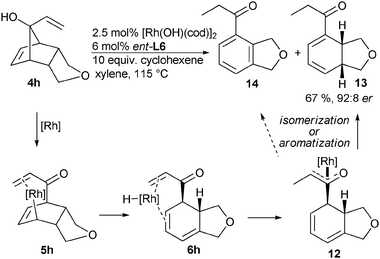 | ||
| Scheme 5 Pathways for the formation of diene 13. | ||
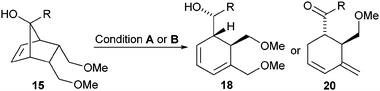
Modifications of the bicyclic framework 1, for example 15 lacking the tetrahydrofuran reveal that subtle differences of the substrate have a significant impact on the reactivity profile of the allyl rhodium species (Scheme 6). In addition, slight modifications of the reaction conditions impact the fate of the putative organometallic species 16 as well.
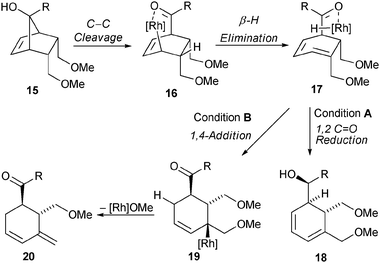 | ||
| Scheme 6 Proposed reaction pathways for the formation of 18 or 20. | ||
For example, performing the retro-allylation of 15 with [Rh(OH)(cod)]2, L6 and caesium carbonate (conditions A), presumably forms regio-isomeric diene 17. The concomitantly formed rhodium hydride species then selectively reduces the carbonyl group of 17 yielding 18 over the previously dominant 1,4-reduction of the diene moiety. In contrast, [Rh(OAc)(C2H4)2]2, L6 and 4 Å molecular sieves instead of caesium carbonate (conditions B) selectively provide diene 20 with an exo-methylene group by the following mechanistic scenario: a 1,4-hydrorhodation of 17 leads to alkyl rhodium species 19, which in turn undergoes β-alkoxide elimination to yield 20.13 A range of secondary alcohols 18 and methylene cyclohexenes 20 can be accessed from norbornenols 15 using these complementary conditions (Table 3). The observed enantioselectivities for the formation of 20 (entries 2, 4, 6, 8) are excellent and generally significantly higher than those for the formation of 18 (entries 1, 3, 5, 7), demonstrating a pronounced effect of the added base on the selectivity.11 The relative configuration of the secondary alcohol function of 18 was assigned by X-ray crystal structure analysis of the Diels–Alder adduct 21 obtained from 18a and 4-phenyl-3H-1,2,4-triazoline-3,5-dione (PTAD) (Scheme 7).§14 This selectivity suggests a facial reduction of the carbonyl group as depicted for 17 (Scheme 5).
| Entry | 15 | Condition | R | 18/20 | % Yieldb | er c |
|---|---|---|---|---|---|---|
| a Conditions A: 0.05 mmol 15, 5 mol% [Rh(OH)(cod)]2, 12.0 mol% ent-L6, 1.0 equiv. Cs2CO3, PhCl (0.15 M), 120 °C, 12 h; Conditions B: 0.05 mmol 15, 5 mol% [Rh(OAc)(C2H4)2]2, 12.0 mol% ent-L6, 20 mg 4 Å MS, PhCl (0.15 M), 120 °C, 12 h. b Isolated product 18 or 20. c Determined by HPLC with a CSP (sign of the optical rotation). d With L6. | ||||||
| 1 | 15a | A | Ph | 18a | 76 | 78∶22 (+) |
| 2 | 15a | B | Ph | 20a | 68 | 95∶5 (−) |
| 3d | 15b | A | 4-MeO–C6H4 | ent-18b | 75 | 88∶12 (−) |
| 4 | 15b | B | 4-MeO–C6H4 | 20b | 82 | 98∶2 (−) |
| 5 | 15c | A | 4-Cl–Ph | 18c | 72 | 89∶11 (+) |
| 6 | 15c | B | 4-Cl–Ph | 20c | 71 | 99∶1 (−) |
| 7d | 15d | A | 3,5-Me–C6H3 | ent-18d | 75 | 88∶12 (−) |
| 8 | 15d | B | 3,5-Me–C6H3 | 20d | 81 | 98∶2 (−) |
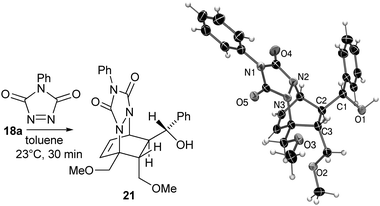 | ||
| Scheme 7 Determination of the relative configuration of 18 as its Diels–Alder adduct 21. | ||
In summary we showed that bicyclic meso-tert-norbornenols can be desymmetrized by retro-allylation mechanisms with chiral rhodium(I) catalysts. Subtle differences of the substrate structure and more importantly of the reaction conditions lead to diverging reaction pathways and hold the promise of a further rich downstream chemistry. Ongoing research is directed towards a deeper understanding and control of the individual steps as well as the development of synthetic applications.
N. C. thanks the Fonds der Chemischen Industrie for a Liebig-Fellowship. M. W. thanks the Roche Research Foundation and the Alexander von Humboldt Foundation for fellowships. We are grateful to Solvias for MeOBiphep, Takasago for Segphos and H8-Binap and Umicore for rhodium salts.
Notes and references
- S. C. Denmark and N. G. Almstead, in Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000, ch. 10, pp. 299–401 Search PubMed.
- For an overview see: (a) H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2009, 82, 778–792 CrossRef CAS; Recent examples with zinc: (b) P. Jones, N. Millot and P. Knochel, Chem. Commun., 1998, 2405 RSC; (c) P. Jones and P. Knochel, Chem. Commun., 1998, 2407 RSC; (d) P. Jones and P. Knochel, J. Org. Chem., 1999, 64, 186 CrossRef CAS; with tin: (e) A. Yanagisawa, T. Aoki and T. Arai, Synlett, 2006, 2071 CrossRef CAS; with gallium: (f) S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2005, 7, 3577 CrossRef CAS; with ruthenium: (g) T. Kondo, K. Kodoi, E. Nishinaga, T. Okada, Y. Morisaki, Y. Watanabe and T. Mitsudo, J. Am. Chem. Soc., 1998, 120, 5587 CrossRef CAS; with nickel: (h) D. Necas, M. Tursky and M. Kotora, J. Am. Chem. Soc., 2004, 126, 10222 CrossRef CAS; (i) Y. Sumida, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 1629 CrossRef CAS.
- (a) S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2006, 128, 2210 CrossRef CAS; (b) Y. Takada, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2006, 8, 2515 CrossRef CAS; (c) S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 12650 CrossRef CAS; (d) M. Iwasaki, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2007, 129, 4463 CrossRef CAS; (e) M. Iwasaki, S. Hayashi, K. Hirano, H. Yorimitsu and K. Oshima, Tetrahedron, 2007, 63, 5200 CrossRef CAS; (f) M. Iwasaki, H. Yorimitsu and K. Oshima, Bull. Chem. Soc. Jpn., 2009, 82, 249 CrossRef CAS; (g) M. Iwasaki, H. Yorimitsu and K. Oshima, Synlett, 2009, 2177 Search PubMed.
- For reviews on enantioselective carbonyl allylation see: (a) J. W. J. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732 CrossRef CAS; (b) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS; (c) I. Marek and G. Sklute, Chem. Commun., 2007, 1683 RSC; (d) D. G. Hall, Synlett, 2007, 1644 CrossRef CAS.
- R. Shintani, K. Takatsu and T. Hayashi, Org. Lett., 2008, 10, 1191 CrossRef CAS.
- M. Waibel and N. Cramer, Angew. Chem., Int. Ed., 2010, 49, 4455 CrossRef CAS.
- L. Xue, K. C. Ng and Z. Lin, Dalton Trans., 2009, 5841 RSC.
- Ring-opening reactions of oxa- and aza-bicyclic compounds follow a distinctively different mechanism. For leading references see: (a) M. Lautens, K. Fagnou and S. Hiebert, Acc. Chem. Res., 2003, 36, 48 CrossRef CAS; (b) M. Lautens and S. Hiebert, J. Am. Chem. Soc., 2004, 126, 1437 CrossRef CAS.
- At least two regioisomeric dienes 6 were observed as transients, but they mostly convert to 8 during the course of the reaction.
- (a) F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6338 CrossRef CAS; (b) F. Shibahara, J. F. Bower and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14120 CrossRef CAS; (c) J. F. Bower, R. L. Patman and M. J. Krische, Org. Lett., 2008, 10, 1033 CrossRef CAS.
- The inorganic base is required to ensure a high reactivity and clean reaction, however at the expense of some enantioselectivity.
- Small, but detectable amounts of 12 are formed during the reaction.
- (a) M. Lautens, C. Dockendorff, K. Fagnou and A. Malicki, Org. Lett., 2002, 4, 1311 CrossRef CAS; (b) L. Navarre, S. Darses and J.-P. Genet, Chem. Commun., 2004, 1108 RSC; (c) T. Miura, T. Sasaki, T. Harumashi and M. Murakami, J. Am. Chem. Soc., 2006, 128, 2516 CrossRef CAS.
- For determination of the absolute configuration of ent-18d see ESI.‡ Retro-allylations with L6 proceed Re with the norbornenol scaffold.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization data. CCDC 779921. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc01950j |
§ Crystallographic data for 21: C25H27N3O5, M = 449.51, triclinic, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 7.9939 (5) Å, b = 11.9064 (9) Å, c = 13.8485 (12) Å, α = 68.816 (3)°, β = 77.480 (4)°, γ = 72.174 (3)°, V = 1161.6 (2) Å3, Z = 2, Dcalc = 1.285 Mg m−3, T = 100 K, reflections collected: 6601, independent reflections: 4009 (Rint = 0.062), R(all) = 0.0818, wR(gt) = 0.1508. CCDC 779921. , a = 7.9939 (5) Å, b = 11.9064 (9) Å, c = 13.8485 (12) Å, α = 68.816 (3)°, β = 77.480 (4)°, γ = 72.174 (3)°, V = 1161.6 (2) Å3, Z = 2, Dcalc = 1.285 Mg m−3, T = 100 K, reflections collected: 6601, independent reflections: 4009 (Rint = 0.062), R(all) = 0.0818, wR(gt) = 0.1508. CCDC 779921. |
| This journal is © The Royal Society of Chemistry 2011 |