Hyperbranched alternating block copolymers using thiol–yne chemistry: materials with tuneable properties†‡
Dominik
Konkolewicz
a,
Cheuk Ka
Poon
a,
Angus
Gray-Weale
b and
Sébastien
Perrier
*a
aKey Centre for Polymers & Colloids, School of Chemistry, University of Sydney, Building F11, NSW 2006, Australia. E-mail: sebastien.perrier@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 3366
bSchool of Chemistry, Monash University, Victoria 3800, Australia
First published on 1st September 2010
Abstract
Alternating-block hyperbranched polymers were synthesized using the highly versatile thiol–yne reaction. Dimethyl acrylamide–styrene and tert-butyl acrylate–styrene polymers were prepared, with subsequent hydrolysis of the tert-butyl ester to acrylic acid. The dimethyl acrylamide–styrene hyperbranched polymers self-assembled into large aggregates, as did the acrylic acid–styrene system at low pH. However, high pH triggers the formation of very well defined small particles in the latter system.
Highly branched polymers such as dendrimers and hyperbranched polymers have many branch points and dense structures, making them candidates for applications such as drug delivery vehicles, viscosity modifiers, supports for catalysts and scaffolds for further synthesis.1–3Dendrimers have regular structures arranged into generations.1 Their well defined structure make dendrimers attractive candidates for applications. However, each generation of a dendrimer must be added sequentially,4–6 making their synthesis challenging. Hyperbranched polymers can be used as alternatives to dendrimers in many applications since these polymers also have many branch points and end-groups.3 Unlike the regular branching in dendrimers, the branch points in a hyperbranched polymer are randomly distributed throughout the polymer. The advantage of hyperbranched polymers is that they can easily be synthesized, often in one pot.3,7 Recently our group developed a new synthesis for hyperbranched polymers based on thiol–yne ‘click’ chemistry.8 This method was used to make hyperbranched polymers from small organic molecules and oligomers of styrene at room temperature. This versatile reaction is unique in that it leads to macromolecules showing near 100% branching points (Scheme 1).8
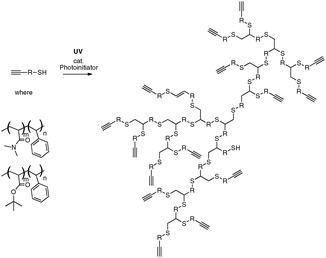 | ||
| Scheme 1 Synthetic scheme for the thiol–yne synthesis of hyperbranched block copolymers. | ||
In this communication we use the thiol–yne approach to make hyperbranched polymers from diblock copolymers. Applying the thiol–yne reaction to block copolymers gives a polymer with alternating blocks in the material. The literature reveals only a small number of branched materials with a block structure,9–12 and very few reports of an alternating block structure.13,14 The latter materials were made using anionic polymerization. This work outlines a facile synthesis of hyperbranched polymers with alternating hydrophobic styrene blocks, and blocks of a hydrophilic monomer, dimethyl acrylamide and acrylic acid. This work also studies the self-assembly properties of these structures in water.
The versatility of the thiol–yne reaction is shown by synthesizing hyperbranched polymers based on linear-diblocks of dimethyl acrylamide (DMAm) and styrene (Sty). The linear chain was synthesized using reversible addition–fragmentation chain transfer (RAFT) polymerization,15,16 with the RAFT agent (prop-2-ynyl propanoate)yl butyl trithiocarbonate (PYPBTC). RAFT was chosen since it is an excellent method for the synthesis of short polymers of well controlled molecular weights and distributions.17 The RAFT synthesis of the linear DMAm block, followed by chain extension with Sty, was performed using typical reaction conditions (see ESI‡). The Mn of the DMAm homopolymer and DMAm–Sty diblock polymers, measured by size exclusion chromatography (SEC), were 620 g mol−1 (PDI = 1.14) and 1600 g mol−1 (PDI = 1.18), respectively, both of which agreed well with theoretical molecular weights, and the block lengths determined by NMR also agreed well with the theoretical ones (see ESI‡). Fig. 1 shows the DMAm and DMAm–Sty SEC traces, with a clear shift of the diblock copolymer to smaller elution volumes, and the absence of low elution volume shoulders on either trace indicates that there is essentially no polymerization through the alkyne on the RAFT agent.
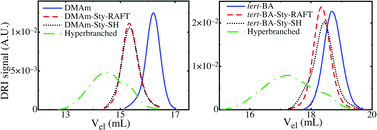 | ||
| Fig. 1 SEC traces of the polymers based on DMAm–Sty (left) and tBA–Sty (right) block copolymers. | ||
The trithiocarbonate on the DMAm–Sty polymer was converted to a thiol by treating the polymer with 50 mol excess of isopropyl amine for 24 h at 25 °C, to yield alkyne–dimethyl acrylamide styrene thiol (ADMAST) polymer. The removal of the trithiocarbonate group was confirmed by the disappearance of the terminal methyl group in the 1H-NMR spectrum after aminolysis. Fig. 1 shows that the SEC trace of ADMAST is similar to the trace before aminolysis, indicating that there is minimal thiol–thiol coupling.
ADMAST was transformed into a hyperbranched polymer by the addition of 5 wt% DMPA photoinitiator in DMF. The reaction mixture was irradiated by UV radiation for 5 hours at room temperature. As seen by SEC (Fig. 1), the hyperbranched polymer molecular weight distribution shifts to much smaller elution volumes, and higher molecular weights, as previously observed for the thiol–yne polymerization of styrene oligomers.8 The final hyperbranched polymer still has some linear chains remaining, however, peak deconvolution shows that these linear chains represent less than 10% of the polymer mass (see ES1‡). In the thiol–yne system studied, the addition of the first thio radical to the alkyne is slower than the addition of the second thio radical to the resulting alkyne.18 This suggests that the thiol–yne reaction gives polymers with very high degrees of branching. Therefore, the thiol–yne reaction gives hyperbranched polymers with alternating blocks of DMAm and Sty. The DMAm–Sty hyperbranched polymer was also characterized by viscometric and DRI SEC, to give a polymer with an Mw of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (PDI = 3.5), indicating the formation of high molecular weight polymers.
000 g mol−1 (PDI = 3.5), indicating the formation of high molecular weight polymers.
The self-assembly of DMAm–Sty hyperbranched polymers was studied in water∶acetone mixtures. Typically linear polymers based on ∼10 units of DMAm and ∼10 units of Sty assemble into micellar structures with hydrodynamic sizes of ∼10 nm (see ESI‡). In contrast, the hyperbranched polymers based on DMAm–Sty do not follow this trend, and in a 20∶80 wt% mixture of acetone∶water these materials form large aggregates with sizes approx. 100 nm. This is seen in Fig. 2a (left vial) which shows significant scattering from the DMAm–Sty sample. In addition Fig. 2b shows the size distribution of the self-assembled DMAm–Sty polymers, with an average size of approx. 70 nm. Another feature seen in Fig. 2b is that the size-distribution of these aggregates shows some run-to-run variation, indicating that the self-assembled structures are not very well-defined. These results suggest that the hyperbranching drastically affects the self-assembly of these DMAm–Sty polymers. If materials that form aggregates are desired, then hyperbranched DMAm–Sty polymers are excellent candidates, however, it may also be desirable to have materials that self-assemble into better defined structures. Such structures can be achieved by altering the lypophilic balance of the blocks in the hyperbranched polymer, and enhancing the hydrophilicity of one block. This can be achieved by introducing a charged group into the polymer, which will improve the material's solubility in water, whilst electrostatic repulsions will limit aggregation.
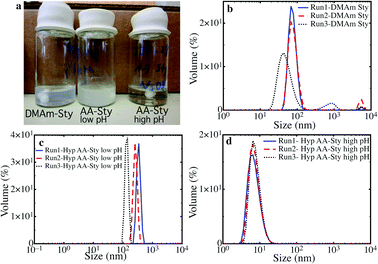 | ||
| Fig. 2 (a) Photograph of self-assembled DMAm–Sty, and AA–Sty based hyperbranched polymers. (b) Size distribution of self-assembled DMAm–Sty hyperbranched polymers. Size distribution of self-assembled AA–Sty hyperbranched polymers at low pH (c) and high pH (d). | ||
In order to synthesize hyperbranched polymers with an ionisable group, the thiol–yne reaction was applied to linear diblock copolymers of tert-butyl acrylate (tBA) and styrene. After polymerization, the tert-butyl group can be removed by hydrolysis to yield the ionisable acrylic acid group.19 Note that, although direct RAFT polymerisation of acrylic acid is possible, tBA was preferred, in order to avoid potential side reactions between the carboxylic acid functionality of the acrylic acid repeating unit and either the amine used for the aminolysis reaction, or the thiol terminal groups. PYPBTC was used to polymerize tBA, giving a well defined linear polymer with a Mn of 1200 g mol−1 (PDI = 1.13), and the molecular weight agrees well with the theoretical molecular weight. The tBA block was chain extended using styrene, giving a polymer with Mn of 1800 g mol−1 (PDI = 1.15), and the block lengths determined by NMR showed acceptable agreement with theory (see ESI‡). Fig. 1 shows the SEC trace of the tBA homopolymer and the tBA–Sty block polymer, and a clear shift of the distribution towards low elution volume is observed after chain extension. In addition, neither the SEC trace of the tBA homopolymer, nor the tBA–Sty polymer show a low elution volume shoulder, indicating well controlled polymers, and essentially no polymerization through the alkyne on the RAFT agent.
This tBA–Sty polymer was converted into a macromonomer for the thiol–yne reaction by transforming the trithiocarbonate group into a thiol, to form alkynetert-butyl acrylate–styrene thiol (AtBAST) polymer. This was achieved by treating the polymer with 50 mol excess of isopropyl amine and stirring for 24 h at 25 °C. The conversion of the trithiocarbonate group to a thiol was confirmed by the disappearance of the methyl group on the trithiocarbonate in the 1H-NMR spectrum after aminolysis. Fig. 1 shows the SEC distribution of the AtBAST polymer, which is very similar to the SEC trace of the polymer before aminolysis, suggesting that there is limited thiol–thiol coupling present in the final product. The AtBAST macromonomer was converted into a hyperbranched polymer by the addition of 5 wt% DMPA photoinitiator and dissolving the mixture in DMF. The reaction mixture was placed under 365 nm UV radiation for 5 hours at room temperature. The SEC trace of the hyperbranched material is also shown in Fig. 1. The SEC distribution of the hyperbranched polymer has clearly shifted to shorter retention times and higher molecular weights. Similar to the DMAm–Sty based hyperbranched polymers there is a residual amount of linear starting material, however, peak deconvolution shows that these linear chains contribute less than 10% of the polymer mass (see ESI‡). As discussed earlier, the kinetics of thiol–yne reaction gives very few intermediate alkene units, and very high degrees of branching.8 This gives rise to hyperbranched polymers with an alternating tBA–Sty structure. The selective hydrolysis of the tert-butyl ester groups to carboxylic acid groups (see ESI‡) yielded hyperbranched polymers with an alternating acrylic acid–styrene (AA–Sty) structure. The AA–Sty hyperbranched polymer was characterized by viscometric and DRI SEC, to give a polymer with an Mw of 37000 g mol−1 (PDI = 7.4), indicating the formation of high molecular weight polymers.
The self-assembly of the hyperbranched polymers with alternating AA–Sty blocks was studied at various pH. At low pH the AA groups are unionized and the material forms large aggregates, similar to the DMAm–Sty hyperbranched polymers. This can be seen in Fig. 2a (middle vial), showing significant scattering from the low pH AA–Sty sample. Fig. 2c shows that the size distribution of these self-assembled materials (average size of approx. 500 nm) is poorly defined, and different runs lead to very different size distributions. These results are similar to the self-assembly of linear AA–Sty polymers at low pH (see ESI‡).
In contrast, at high pH the AA groups are ionized and the material self-assembles into a much smaller structures. This is illustrated by Fig. 2a (right vial), where the AA–Sty sample at high pH leads to a transparent solution. This observation was confirmed by light scattering analyses (Fig. 2d), which revealed a size distribution of the AA–Sty system at high pH of approximately 10 nm. The self-assembly of AA–Sty at high pH is well defined, and multiple runs lead to the same distribution. Similar sizes are seen in self-assembled linear AA–Sty polymers (see ESI‡).
At high pH the hyperbranched structures carry a large charge, which causes significant electrostatic repulsion between molecules. This suggests that the material could self-assemble into unimolecular particles to reduce electrostatic repulsions, while still providing minimal contact between the styrene segments and the aqueous phase. These unimolecular particles are consistent with the particle size measured, and should have the styrene units at the center, with the charged groups forming loops between the styrene units. Although the linear AA–Sty chains self-assemble into structures with similar sizes to the hyperbranched polymer at high pH, the self-assembly of the linear chain contains many molecules, whereas the hyperbranched polymer could form a unimolecular particle. We are currently investigating further the structure of these particles. Another factor affecting the self-assembly is the hydrophilicity of the blocks. The difference in structure obtained when varying the hydrophilic segment from dimethyl amide to carboxylate suggests that the carboxylate is more water soluble than the amide. This argument implies that polymers with the carboxylate groups are likely to self-assemble into small structures, since a small structure will allow more contact between carboxylate groups and water. The drive to form small particles should be weaker for the less hydrophilic amide and carboxylic acid group.
This work extended the concept developed by our group, where molecules with thiols and alkynes are used to synthesize hyperbranched polymers at room temperature. In this work, we demonstrated the versatility of the thiol–yne reaction by applying it to block copolymers as repeating units. Linear block copolymers were used as macromonomers to form hyperbranched polymers with an alternating block structure. In addition to synthesizing hyperbranched alternating block polymer, we also studied their self-assembly properties. We found that hyperbranched block copolymers based on dimethyl acrylamide and styrene gave rise to large aggregates in water acetone mixtures, while acrylic acid based polymers formed very large aggregates when the acid group is unionized, but gave well defined small particles when the acid groups were ionized. These results show the possibility of using the thiol–yne synthesis to make hyperbranched polymers with interesting structures and tuneable properties. Due to the self-assembly properties of these hyperbranched materials, they could be used in drug delivery and other applications.
We are grateful to Meiliana Siauw for providing linear AA–Sty polymers.
Notes and references
- F. Aulenta, W. Hayes and S. Rannard, Eur. Polym. J., 2003, 39, 1741 CrossRef CAS.
- J. M. J. Fréchet, C. J. Hawker, I. Gitsov and J. W. Leon, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 33, 1399 Search PubMed.
- M. Jikei and M.-a. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233 CrossRef CAS.
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS.
- G. R. Newkome, Z. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003 CrossRef CAS.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J. (Tokyo), 1985, 17, 117 Search PubMed.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075 CrossRef CAS.
- M. Trollsas, M. A. Kelly, H. Claesson, R. Siemens and J. L. Hedrick, Macromolecules, 1999, 32, 4917 CrossRef.
- D. E. Lonsdale, M. R. Whittaker and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6292 CrossRef CAS.
- C. N. Urbani, C. A. Bell, D. Lonsdale, M. R. Whittaker and M. J. Monteiro, Macromolecules, 2008, 41, 76 CrossRef CAS.
- Y. Zhou and D. Yan, Chem. Commun., 2009, 1172 RSC.
- L. R. Hutchings, Soft Matter, 2008, 4, 2150 RSC.
- L. R. Hutchings, J. M. Dodds, D. Rees, S. M. Kimani, J. J. Wu and E. Smith, Macromolecules, 2009, 42, 8675 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715 CrossRef CAS.
- D. Konkolewicz, M. Siauw, A. Gray-Weale, B. S. Hawkett and S. Perrier, J. Phys. Chem. B, 2009, 113, 7086 CrossRef CAS.
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113 CrossRef CAS.
- K. A. Davis and K. Matyjaszewski, Macromolecules, 2000, 33, 4039 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental, additional characterization of DMAm–Sty, tBA–Sty, AA–Sty polymers, self-assembly of linear polymers. See DOI: 10.1039/c0cc02429e |
| This journal is © The Royal Society of Chemistry 2011 |