A solid-state trimerisation of a diene diacid affords a bicyclobutyl: reactant structure from X-ray powder data and product separation and structure determination viaco-crystallisation†‡
Manza B. J.
Atkinson
a,
Ivan
Halasz
b,
Dejan-Krešimir
Bučar
a,
Robert E.
Dinnebier
b,
S. V.
Santhana Mariappan
a,
Anatoliy N.
Sokolov
a and
Leonard R.
MacGillivray
*a
aDepartment of Chemistry, University of Iowa, Iowa City, Iowa, USA. E-mail: len-macgillivray@uiowa.edu; Fax: +1 319-335-1270; Tel: +1 319-335-3504
bMax-Planck-Institute for Solid State Research, Heisenbergstr. 1, 70569 Stuttgart, Germany
First published on 31st August 2010
Abstract
A bicyclobutyl that bears six carboxylic acid groups results from a trimerisation of a diene diacid in the solid state. Powder X-ray diffraction and a co-crystallisation are used to solve the structure of the diene and elucidate the stereochemistry of the bicyclobutyl, respectively.
The solid state continues to attract attention as a medium for the synthesis of organic molecules.1 Crystalline solids are dominated by intermolecular forces that restrict molecular movement and, as a result, can provide solvent-free access to products that may otherwise be unavailable in solution.2 Whereas chemists are learning how to utilise noncovalent bonds, in the form of supramolecular synthons,3 to assist arrangements of molecules in solids into positions for reaction (e.g. templates, hosts, salts),4 reactions that occur in the solid state without assistance of supramolecular synthesis can also afford important products.5,6 Moreover, with the significance of recent advancements in crystal structure determination via single-crystal (i.e. rapid data collection using CCDs) and powder X-ray diffraction (PXRD) now being realised, it is becoming apparent that the extent to which reactions that occur in the solid state can impact organic synthetic chemistry is yet to be fully realised and developed.
In this context, bicyclobutyl is a strained molecule composed of two cyclobutane rings connected at two corners via a carbon–carbon single (C–C) bond (Scheme 1). The bicyclobutyl framework has recently emerged as an alternative high-energy hydrocarbon component of propellants and fuels.7 Chemists have typically employed solution-based methods to synthesize bicyclobutyls.8 Transformations based on metal-mediated couplings using Grignard and lithium reagents, as well as free radical and anionic-initiated polymerizations of functionalized cyclobutanes, have been reported.
![(a) Bicyclobutyl and (b) solid-state [2+2] photodimerisation of three dienes to generate a bicyclobutyl.](/image/article/2011/CC/c0cc02204g/c0cc02204g-s1.gif) | ||
| Scheme 1 (a) Bicyclobutyl and (b) solid-state [2+2] photodimerisation of three dienes to generate a bicyclobutyl. | ||
In principle, a simple means to form bicyclobutyl is to arrange three dienes in a geometry for two intermolecular [2+2] photodimerisations (Scheme 1). A trimerisation of this type would be expected to bear a high energetic cost in solution owing to effects of solvation and entropy. Given that the [2+2] photodimerisation readily proceeds in solids, the restricted yet flexible environment of the solid state has an inherent capacity to circumvent problems of solvation and entropy to make a trimerisation of a diene possible.9
Herein, we report that the diene (E,E)-2,5-dimethylmuconic acid 1 undergoes a trimerisation in the solid state to afford the bicyclobutyl 2a (Scheme 2). Our interests in 1 lie in the ability of the diacid to undergo an intermolecular [2+2] photodimerisation to give a [3]-ladderane as a product. During studies to use hydrogen-bond-acceptor templates to assemble 1 for photoreaction,1 we have discovered that pure 1 undergoes a trimerisation in the solid state to generate the bicyclobutyl hexaacid diene 2a, as well as the related anti dimer 2b. To understand the origin of the photoreactivity, and to circumvent difficulties encountered to grow single crystals of 1, we have turned to powder X-ray diffraction to determine the solid-state packing of the diene.§ Additionally, difficulties to isolate and grow single crystals of the photoproduct have prompted us to apply the rapidly-developing process of co-crystallisation to separate 2a from 2b and confirm the stereochemistry of the bicyclobutyl by single-crystal X-ray diffraction.¶ Whereas olefinic mono- (e.g.cinnamic acid) and dicarboxylic acids (e.g.fumaric acid) have been central to understanding the origins of the [2+2] photodimerisation in the solid state,5,101 represents, to our knowledge, the first case wherein an olefinic carboxylic acid undergoes a trimerisation to form a bicyclobutyl in a solid.11
 | ||
| Scheme 2 Solid-state photodimerisation of 1 to give 2a and 2b. | ||
When crystalline 1 was subjected to broadband UV-irradiation (450 W medium pressure Hg lamp) for a period of ca. 60 h, a 1H NMR spectrum (DMSO-d6) revealed the diene to be consumed in a [2+2] photodimerisation in near quantitative yield (Fig. 1). A photoreaction was evidenced by the near complete disappearance of the olefinic protons at 7.23 ppm and the emergence of three peaks, in the form of two doublets and a singlet at 4.21, 4.04, and 3.35 ppm in the cyclobutane region, respectively. The signal for the methyl protons also disappeared and was replaced by five singlets in the region of 1.21–1.82 ppm, while two new peaks, in the form of two doublets, appeared in the vinyl region at 5.85 and 5.72. The NMR data were also consistent with the photoreaction having generated two products. The formation of two products was evidenced by a 1H–1H COSY spectrum, which revealed two groups of resonances in the olefin region that correlated as two separate J-coupling networks in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 ratio (ESI†). An electrospray ionization mass spectrum also showed two major peaks at m/z 339.11 and 509.17, which suggested the diacid reacted to give a trimer and dimer (ESI†). From the combined NMR and mass spectrometry data, we concluded that the solid-state reaction afforded the bicyclobutyl trimer 2a (35% yield) and the anti photodimer of 2b (65% yield). A diagnostic peak in the NMR spectrum for the trimer is the lone cyclobutane singlet at 3.35 ppm, which is assigned to the two H-atoms that span the C–C bond that connect the cyclobutane rings of 2a. Assignment of the peak to the network of 2a was also verified by single-bond and multiple-bond 13C–1H correlation data (HMBC) of the photoreacted product mixture (ESI†). A NOE experiment also demonstrated a close interaction between the methyl and olefinic protons in 2b, which is consistent with the anti geometry (ESI†).
∶1 ratio (ESI†). An electrospray ionization mass spectrum also showed two major peaks at m/z 339.11 and 509.17, which suggested the diacid reacted to give a trimer and dimer (ESI†). From the combined NMR and mass spectrometry data, we concluded that the solid-state reaction afforded the bicyclobutyl trimer 2a (35% yield) and the anti photodimer of 2b (65% yield). A diagnostic peak in the NMR spectrum for the trimer is the lone cyclobutane singlet at 3.35 ppm, which is assigned to the two H-atoms that span the C–C bond that connect the cyclobutane rings of 2a. Assignment of the peak to the network of 2a was also verified by single-bond and multiple-bond 13C–1H correlation data (HMBC) of the photoreacted product mixture (ESI†). A NOE experiment also demonstrated a close interaction between the methyl and olefinic protons in 2b, which is consistent with the anti geometry (ESI†).
 | ||
| Fig. 1 1H NMR spectrum of 1 after photoreaction (T = 2a; D = 2b). | ||
To understand the reactivity of 1, we attempted to grow single crystals of the diacid. Although the diacid was soluble in common polar and apolar solvents, conventional methods of crystallisation (e.g. supersaturation, slow cooling) afforded microcrystalline powders. Consequently, we turned to powder X-ray diffraction (PXRD) to elucidate the structure of 1. PXRD has received increasing attention as a means to extract structural information from organic powder samples.12–14 When a high-resolution powder diffraction pattern was collected on 1, the data indexed to give a monoclinic unit cell with P21/c as the most likely space group. According to volume increments, it was determined that the unit cell contained two molecules of 1. The crystal structure was then solved by direct space global optimisation assuming the asymmetric unit to consist of one half of molecule of 1 and confirmed by refinement according to the Rietveld method (ESI†).
The diene was determined from the PXRD data to self-assemble (Fig. 2), similar to the parent muconic acid,15 in a one-dimensional (1D) hydrogen-bonded polymer sustained by carboxylic-acid dimers (O–H⋯O distance: 2.649(2) Å) (Fig. 2a). The polymers lie stacked offset interacting via face-to-face π⋯π interactions along the crystallographic a-axis. Importantly, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of nearest-neighbour dienes lie approximately parallel such that each CC bond sits in close proximity to two adjacent CC bonds at separations of 3.79 Å and 3.89 Å (Fig. 2b). The geometry of 1 satisfies the criteria of Schmidt for a photodimerisation,5 wherein the diene can react to give the trimer 2a, the related photodimer 2b, as well as higher oligomers and polymers.
C bonds of nearest-neighbour dienes lie approximately parallel such that each CC bond sits in close proximity to two adjacent CC bonds at separations of 3.79 Å and 3.89 Å (Fig. 2b). The geometry of 1 satisfies the criteria of Schmidt for a photodimerisation,5 wherein the diene can react to give the trimer 2a, the related photodimer 2b, as well as higher oligomers and polymers.
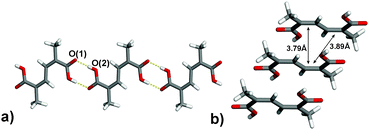 | ||
| Fig. 2 X-ray crystal structure of 1 as determined by PXRD: (a) hydrogen-bonded polymer and (b) stacked CC bonds along a-axis. | ||
 | ||
| Fig. 3 X-ray structure of (2a)·(4,4′-dipy)·2DMF: (a) trimer 2a and (b) 1D hydrogen-bonded assembly showing the stacked bipyridines. | ||
Importantly, the structure from the PXRD experiment is consistent with three molecules of 1 having reacted in a trimerisation to afford 2a. In such a transformation, the diene undergoes two intermolecular [2+2] photodimerisations that produce a bicyclobutyl composed of eight stereocenters, six carboxylic acid groups, and two unreacted CC bonds. In addition to 2a, the packing is consistent with 1 having reacted to give the anti photodimer of 2b. The shortest CC separation (3.79 Å) and a θ value of 96.5° place the p-orbitals6 in a favourable position to react to give anti2b. The resulting photodimer presumably serves as a precursor in a second cycloaddition that reacts to give the trimer.
Whereas attempts to separate the trimer 2a and dimer 2b using chromatography were unsuccessful, 2a was cleanly separated from 2b and structurally characterized via a co-crystallisation. Given that both photoproducts are decorated with acid groups, we hypothesized that co-crystallisation of the crude reacted solid mixture with a co-crystal former (CCF) lined with hydrogen-bond-acceptor functionality could be used to isolate and, possibly, confirm the structure of a photoproduct via a selective crystallisation. A co-crystallisation would be promoted by the specificity of the hydrogen bonds between the CCF and photoproduct, as well as crystal packing. Co-crystallisations have been employed, in relatively few cases, to determine molecular structure16 while an application to isolate a natural product has recently been reported.17
When a sample of the reaction mixture (100.0 mg) from 1 was combined with 4,4′-dipyridyl (4,4′-dipy) (21.0 mg) in DMF (1.0 mL) and allowed to sit for a period of one week, colourless single crystals suitable for X-ray diffraction were obtained. A 1H NMR spectrum (DMSO-d6) revealed signals that matched 2a in the form of (2a)·2(4,4′-dipy)·2DMF.
An X-ray analysis of (2a)·2(4,4′-dipy)·2DMF confirmed the structure of 2a (Fig. 3a). The bicyclobutyl 2a sits around a centre of inversion that is positioned at the central C–C bond [C–C distance (Å): 1.523(2)] with the C–C distances of the cyclobutane rings comparing well to experimentally-related structures [C–C distances (Å): C(2)–C(3) 1.584(2), C(3)–C(4) 1.575(2), C(4)–C(5) 1.574(2), C(5)–C(2) 1.579(2)].18 The components self-assemble to form a 1D hydrogen-bond chain, which runs approximately parallel to the crystallographic c-axis, held together by O–H⋯N hydrogen bonds [O⋯N distances (Å): O(1)⋯N(1) 2.667(2), O(4)⋯N(2) 2.685(2)] (Fig. 3b). The four inner carboxylic acid groups (i.e. across cyclobutane rings) of 2a participate in the hydrogen bonds to 4,4′-dipy with the bipyridine being propagated as stacked dimers along the chain. Each outer acid group participates in an O–H⋯O hydrogen bond to an included DMF molecule [O⋯O distance (Å): O(6)⋯O(7) 2.624(2)]. The co-crystal (2a)·2(4,4′-dipy)·2DMF represents, to our knowledge, the first structure confirmation of a bicyclobutyl obtained from the solid state.9,15 Efforts are underway to employ a similar approach to elucidate the structure of 2b.
In summary, we have described a solid-state trimerisation of a diene diacid that affords a bicyclobutyl, as well as a related photodimer. An understanding of the origin of the trimerisation has been provided by PXRD while the structure of the trimer has been authenticated viaco-crystallisation. We are currently studying the reactivity properties of the hexaacid with an aim to reduce the bicyclobutyl 2a to hydrocarbon groups. The approach described here may also be applied to additional dienes, and related polyenes, that react to form higher oligomers in solids.
We thank the National Science Foundation (LRM, DMR-0133138) and the Petroleum Research Fund of the American Chemical Society (Type AC Grant) for financial support.
Notes and references
- L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280 CrossRef CAS.
- A. Matsumoto, Top. Curr. Chem., 2005, 254, 263 CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342 CrossRef CAS.
- (a) Y. Ito, B. Borecka, J. Trotter and J. R. Scheffer, Tetrahedron Lett., 1995, 36, 6083 CrossRef CAS; (b) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS.
- (a) G. M. Schmidt, Pure Appl. Chem., 1971, 27, 647 CAS; (b) J. M. Thomas, Philos. Trans. R. Soc. London, Ser. A, 1974, 277, 251 CAS.
- V. Ramamurthy and K. Venkatesan, Chem. Rev., 1987, 87, 433 CrossRef CAS.
- (a) P. E. Eaton, R. L. Gilardi and M.-X. Zhang, Adv. Mater., 2000, 12, 1143 CrossRef CAS; (b) M. V. Savos'kin, L. M. Kapkan, G. E. Vaiman, A. N. Vdovichenko, O. A. Gorkunenko, A. P. Yaroshenko, A. F. Popov, A. N. Mashchenko, V. A. Tkachev, M. L. Voloshin and Y. F. Potapov, Russ. J. Appl. Chem., 2007, 80, 31 CrossRef CAS.
- H. Hopf, J. F. Liebman and H. M. Perks, Cubanes, fenestranes, ladderanes, prismanes, staffanes, and other oligocyclobutanoids in Chemistry of Cyclobutanes, Wiley, 2005, pp. 1061–1109 Search PubMed.
- For a report that suggests such a trimerisation, see: Y. Mori and A. Matsumoto, Chem. Lett., 2007, 36, 510 Search PubMed.
- (a) T. Friščić and L. R. MacGillivray, Chem. Commun., 2005, 5748 RSC; (b) X. Mei, S. Liu and C. Wolf, Org. Lett., 2007, 9, 2729 CrossRef CAS.
- For related oligomerisations, which include trimer formations, see: (a) L. Addadi and M. Lahav, J. Am. Chem. Soc., 1978, 100, 2838 CrossRef CAS; (b) M. Hasegawa, Pure Appl. Chem., 1986, 58, 1179 CAS; (c) M. J. Vela, B. B. Snider, B. M. Foxman, V. Buchholz and V. Enkelmann, Chem. Commun., 2000, 2225 RSC.
- (a) K. D. M. Harris, M. Tremayne, P. Lightfoot and P. G. Bruce, J. Am. Chem. Soc., 1994, 116, 3543 CrossRef CAS; (b) K. D. M. Harris, M. Tremayne and B. M. Kariuki, Angew. Chem., Int. Ed., 2001, 40, 1626 CrossRef CAS; (c) K. D. M. Harris, Cryst. Growth Des., 2003, 3, 887 CrossRef CAS; (d) K. D. M. Harris and E. Y. Cheung, Chem. Soc. Rev., 2004, 33, 526 RSC.
- W. I. F. David and K. Shankland, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 52 CAS.
- (a) S. Karki, L. Fábián, T. Friščić and W. Jones, Org. Lett., 2007, 9, 3133 CrossRef CAS; (b) C. W. Padgett, H. D. Arman and W. T. Pennington, Cryst. Growth Des., 2007, 7, 367 CrossRef CAS; (c) F. Guo, J. Marti-Rujas, Z. Pan, C. E. Hughes and K. D. M. Harris, J. Phys. Chem. C, 2008, 112(50), 19793 CrossRef CAS.
- M. Lahav and G. M. J. Schmidt, J. Chem. Soc. B, 1967, 312 RSC.
- (a) P. M. Bhatt and G. R. Desiraju, CrystEngComm, 2008, 10, 1747 RSC; (b) P.-H. Liu, L. Li, J. A. Webb, Y. Zhang and N. S. Goroff, Org. Lett., 2004, 6, 2081 CrossRef CAS; (c) W. L. Duax, C. Eger, S. Pokrywiecki and Y. Osawa, J. Med. Chem., 1971, 14, 295 CrossRef CAS; (d) C. Eger and D. A. Norton, Nature, 1965, 208, 997 CrossRef CAS.
- M. Khan, V. Enkelmann and G. Brunklaus, J. Am. Chem. Soc., 2010, 132, 5254 CrossRef CAS.
- (a) Y. Kai, P. Knochel, S. Kwiatkowski, J. D. Dunitz, J. F. M. Oth, D. Seebach and H. O. Kalinowski, Helv. Chim. Acta, 1982, 65, 137 CAS; (b) Y.-L. Lam, L.-L. Koh and H.-H. Huang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 397 CrossRef.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Details of 1H NMR studies and structure solutions by single-crystal and powder XRD. CCDC 780278 (1) and 780279 (2a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cc02204g |
| § Crystal data for 1: C8H10O4, Mr = 170.17, monoclinic, a = 3.8984(1) Å, b = 7.8794(1) Å, c = 13.5148(4) Å, β = 95.965(2)°, V = 412.88(1) Å3, T = 293(2) K, space groupP21/c, Z = 2, Rp = 0.0282, Rwp = 0.0376, goodness of fit was 1.72, Rexp = 0.0219, RBragg = 1.40 (CCDC 780278). |
¶ Crystal data for 2a: C24H30O12·2(C10H8N2)·2(C3H7NO), Mr = 969.04, triclinic, a = 8.1654(9) Å, b = 11.7147(13) Å, c = 13.1518(14) Å, α = 86.292(5)°, β = 77.546(5)°, γ = 78.982(5)°, V = 1205.4(2) Å3, T = 150(2) K, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 1, μ(MoKα) = 0.098 mm−1, 8199 reflections measured, 4236 independent reflections (Rint = 0.0182). R1 = 0.0462 (I > 2σ(I)), wR(F2) = 0.1239 (I > 2σ(I)), R1 = 0.0576 (all data), wR(F2) = 0.1294 (all data), goodness of fit on F2 was 1.051 (CCDC 780279). , Z = 1, μ(MoKα) = 0.098 mm−1, 8199 reflections measured, 4236 independent reflections (Rint = 0.0182). R1 = 0.0462 (I > 2σ(I)), wR(F2) = 0.1239 (I > 2σ(I)), R1 = 0.0576 (all data), wR(F2) = 0.1294 (all data), goodness of fit on F2 was 1.051 (CCDC 780279). |
| This journal is © The Royal Society of Chemistry 2011 |