Direct carbon–carbon bond formation via soft enolization: aldol addition of α-halogenated thioesters†‡
Julianne M.
Yost
,
Rachel J.
Alfie
,
Emily M.
Tarsis
,
Insun
Chong
and
Don M.
Coltart
*
Department of Chemistry, Duke University, Durham, NC, USA. E-mail: don.coltart@duke.edu
First published on 21st October 2010
Abstract
α-Halo thioesters undergo soft enolization and syn-selective direct aldol addition to aldehydes in the presence of MgBr2·OEt2 and i-Pr2NEt to produce α-halo-β-hydroxy thioesters.
α-Halo-β-hydroxy carboxylic acid derivatives are useful intermediates in the synthesis of natural products. They are normally generated viaaldol addition of a pre-formed enolate and an aldehyde.1 While effective, the step-wise procedures required to generate the enolates are time consuming, particularly if trapping is involved, and require that all manipulations be conducted under anhydrous conditions and, when strong bases are used, at low temperature. In contrast, soft enolization2,3 provides a mild and straightforward approach to conducting enolate chemistry. Here, rather than forcing deprotonation with a strong base such as LDA, a relatively weakly basic amine is used in combination with a Lewis-acid to effect deprotonation. We have been investigating this mode of enolization with thioesters in the development of direct versions of certain fundamental carbon–carbon bond-forming reactions.4 Given the efficiency and operational simplicity of these transformations, we sought to extend this approach to the synthesis of α-halo-β-hydroxy carboxylate derivatives. In what follows, we describe the development of a MgBr2·OEt2-promoted direct aldol addition reaction between α-halo thioesters and aldehydes using soft enolization, which produces α-halo-β-hydroxy thioesters.
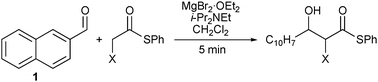
A practical concern associated with the proposed transformation is the possibility of competing Darzens reaction1a,b,d,5 to produce the corresponding α,β-epoxy thioesters (Scheme 1). However, our previous experiences led us to believe that the magnesium aldolate intermediate would be sufficiently stable under the reaction conditions to prevent epoxide formation. To test this, we attempted the aldol addition with aldehyde 1 and α-bromo thioester 2 under our soft enolization conditions4 (Scheme 2). Gratifyingly, the desired α-bromo-β-hydroxy thioester (3) was formed rapidly and in excellent yield, with no indication of epoxide formation.
 | ||
| Scheme 1 Possible reaction pathways. | ||
 | ||
| Scheme 2 MgBr2·OEt2-promoted direct aldol reaction of α-bromo phenylthioacetate (2) with 2-naphthaldehyde (1). | ||
We next examined the effect of the halogen substituent on the reaction. To do this, thioesters 2, 4, and 5 were combined with 1 and allowed to react for 5 min at room temperature, before quenching with acid (Table 1). In each case the desired product was produced in high yield, with α-chloro thioester 4 giving the best result. No appreciable difference in the diastereoselectivity as a function of the halogen was seen. Given the rapid nature of the transformations, control experiments were carried out with thioesters 2 and 4 in which the magnesium salt was omitted from the reaction mixture, but all other components were retained. After 72 hours, no aldol addition product was detected for the reaction involving 2, and only a trace (<5%) was formed when thioester 4 was used, confirming the importance of MgBr2·OEt2.
Having established the superiority of the α-chloro thioester in the addition reaction, we turned our attention to improving the diastereoselectivity. In pioneering work on the development of an anti-selective aldol addition, Heathcock and Pirrung showed that increasing the steric bulk of the ester component led to an increase in diastereoselectivity.6 Thus, various α-chloro thioesters derived from more sterically-demanding thiols were examined (Table 2). As with the previous study, an increase in steric bulk did correlate to an increase in diastereoselectivity. However, in contrast, the syn—not the anti—diastereomer was preferentially formed. Interestingly, a somewhat lower syn selectivity resulted for the more bulky α-chloro thioester 10 than for 9.

| Entry | Thioester | Product | Time/h |
syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶anti ∶anti |
Conversion (%) |
|---|---|---|---|---|---|
| a 1 molar equiv. of 1, 1.2 molar equiv. of thioester, and 1.4 molar equiv. of MgBr2·OEt2 (concn 0.2 M), followed by addition of 2.0 molar equiv. of i-Pr2NEt at rt. | |||||
| 1 | 4 R1 = Ph | 6 | 0.5 | 1.2∶1 |
98 |
| 2 |
8 R1 =  |
11 | 0.5 | 2.5∶1 |
97 |
| 3 |
9 R1 =  |
12 | 0.5 | 5.2∶1 |
97 |
| 4 |
10 R1 = 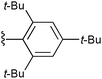 |
13 | 1 | 4.5∶1 |
90 |
With effective conditions in place, the scope of the reaction was explored using thioester 9 and a variety of aldehydes (Table 3). The transformation proceeded efficiently with aromatic aldehydes, including electron rich and deficient systems, and also proceeded well with the highly sterically-hindered aldehyde 18. Notably, when the reaction was carried out with an enolizable aldehyde possessing a single α-proton (19, entry 7), the aldol product was produced in good yield. Encouraged by this result, aldehydes 20 and 21 were tested in the addition reaction. Unfortunately, while the desired products did form (28 and 29, respectively), they were obtained in a relatively low yield due to competing aldehyde self addition.

| Entry | Aldehyde | Product | Time/h |
syn∶anti |
Yield (%) |
|---|---|---|---|---|---|
| a 1 molar equiv. of aldehyde, 1.2 molar equiv. of 9, and 1.4 molar equiv. of MgBr2·OEt2 (concn 0.2 M), followed by addition of 2.0 molar equiv. of i-Pr2NEt at rt. | |||||
| 1 |
1 R1 =  |
12 | 0.5 | 5.2∶1 |
91 |
| 2 |
14 R1 =  |
22 | 0.5 | 4.0∶1 |
96 |
| 3 |
15 R1 =  |
23 | 1 | 4.0∶1 |
96 |
| 4 |
16 R1 = 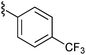 |
24 | 0.5 | 3.7∶1 |
97 |
| 5 |
17 R1 = 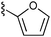 |
25 | 0.5 | 4.2∶1 |
83 |
| 6 |
18 R1 = 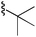 |
26 | 1 | 3.2∶1 |
76 |
| 7 |
19 R1 =  |
27 | 1 | 3.2∶1 |
73 |
| 8 |
20 R1 =  |
28 | 1 | 3.4∶1 |
29 |
| 9 |
21 R1 =  |
29 | 1 | 5.0∶1 |
36 |
In conclusion, we have developed a mild and efficient MgBr2·OEt2-promoted direct aldol reaction of α-chloro thioesters employing soft enolization. The transformation proceeds without competing Darzens addition, producing the α-chloro-β-hydroxy thioesters in moderate to high yields with moderate to good diastereoselectivity. The reaction is effective in the case of an aldehyde having a single enolizable proton. Further studies will address the adaptation of this method to the use of aldehydes having two enolizable protons.
Notes and references
- (a) J. Nebot, P. Romea and F. Urpi, J. Org. Chem., 2009, 74, 7518 CrossRef CAS; (b) S.-i. Kiyooka and K. A. Shahid, Tetrahedron: Asymmetry, 2000, 11, 1537 CrossRef CAS; (c) Y.-C. Wang, D.-W. Su, C.-M. Lin, H.-L. Tseng, C.-L. Li and T.-H. Yan, Tetrahedron Lett., 1999, 40, 3577 CrossRef CAS; (d) E. J. Corey and S. Choi, Tetrahedron Lett., 1991, 32, 2857 CrossRef CAS; (e) D. A. Evans, E. B. Sjogren, A. E. Weber and R. E. Conn, Tetrahedron Lett., 1987, 28, 39 CrossRef CAS.
- For pioneering applications of soft enolization in direct carbon–carbon bond formation see: (a) M. W. Rathke and P. J. Cowan, J. Org. Chem., 1985, 50, 2622 CrossRef CAS; (b) M. W. Rathke and M. Nowak, J. Org. Chem., 1985, 50, 2624 CrossRef CAS; (c) R. E. Tirpak, R. S. Olsen and M. W. Rathke, J. Org. Chem., 1985, 50, 4877 CrossRef CAS.
- D. Lim, G. Zhou, A. E. Livanos, F. Fang and D. M. Coltart, Synthesis, 2008, 2148 CAS.
- See, for example: (a) G. Zhou, D. Lim and D. M. Coltart, Org. Lett., 2008, 10, 3809 CrossRef CAS; (b) J. M. Yost, M. R. Garnsey, M. C. Kohler and D. M. Coltart, Synthesis, 2009, 56 CAS; (c) D. Lim, F. Fang, G. Zhou and D. M. Coltart, Org. Lett., 2007, 9, 4139 CrossRef CAS; (d) J. M. Yost, G. Zhou and D. M. Coltart, Org. Lett., 2006, 8, 1503 CrossRef CAS.
- (a) T. Rosen, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol. 2, pp. 409–439 Search PubMed; (b) A. K. Ghosh and J.-H. Kim, Org. Lett., 2004, 6, 2725 CrossRef CAS; (c) L. N. Pridgen, A. F. Abdel-Magid, I. Lantos, S. Shilcrat and D. S. Eggleston, J. Org. Chem., 1993, 58, 5107 CrossRef CAS; (d) A. Abdel-Magid, L. N. Pridgen, D. S. Eggleston and I. Lantos, J. Am. Chem. Soc., 1986, 108, 4595 CrossRef CAS.
- (a) M. C. Pirrung and C. H. Heathcock, J. Org. Chem., 1980, 45, 1727 CrossRef; (b) H. C. Heathcock, M. C. Pirrung, S. H. Montgomery and J. Lampe, Tetrahedron, 1981, 37, 4087 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and analytical data for compounds 2, 4, 5, 8–10, 12, 22–29. See DOI: 10.1039/c0cc02345k |
| This journal is © The Royal Society of Chemistry 2011 |