Direct and efficient N-heterocyclic carbene-catalyzed hydroxymethylation of aldehydes†‡
Nadine
Kuhl
and
Frank
Glorius
*
Organisch-Chemisches Institut der Westfälischen Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de; Fax: +49 251 8333202; Tel: +49 251 8333248
First published on 1st November 2010
Abstract
The N-heterocyclic carbene-catalyzed coupling of several aldehydes with paraformaldehyde is reported, directly providing the corresponding valuable hydroxymethyl ketones. Results of first mechanistic experiments for this remarkably selective transformation are also provided.
The hydroxymethyl ketone moiety is an important motif of synthetic precursors and biologically active compounds.1 Consequently, many methods for its preparation have been reported. However, most of these methods are rather simple hydrolyses2 or oxidations3 and do not form any new C–C bonds.
An attractive alternative strategy is based on the N-heterocyclic carbene (NHC)4 catalyzed umpolung of aldehydes by the formation of the Breslow-intermediate and the addition of this nucleophilic species to electrophiles as found in the well-known Benzoin condensation5,6 and related reactions.5,7,8 In these transformations, many selectivity issues arise and the recent interest into and progress in the field of NHCs has allowed the development of new, efficient and highly selective processes. In this respect, the NHC-catalyzed addition of aldehydes to formaldehyde seems to be straightforward and an efficient route for the formation of hydroxymethyl ketones. Thus, it comes as a surprise that this strategy has only rarely been applied.9,10 In these remarkable pioneering reports by Inoue et al.9 using an NHC-organocatalyst and Demir et al.10a and Müller et al.10b employing an enzyme as the catalyst, the validity of this approach was impressively shown. However, all of these reports have significant drawbacks, including low isolated yields (ranging from 6–17% for the work of Inoue et al.9) or high dilution (<0.02 M10a), a large excess of formaldehyde (e.g. 32 eq.10a) or long reaction time (e.g. 4 days10a).
An improved preparative method that builds up hydroxymethyl ketones from readily available aldehydes and formaldehyde, increasing the complexity of the carbon skeleton, would be highly desirable. Herein, we report the NHC-catalyzed direct coupling of aldehydes with formaldehyde resulting in hydroxymethyl ketones in good isolated yields.
We commenced our investigation with the hydroxymethylation of benzaldehyde, employing paraformaldehyde (Table 1) as the CH2O source. Since in many recent studies the choice of NHC has been crucial,5 we started off by screening different classes of NHC-organocatalysts (entries 1–6). Whereas imidazolylidenes and benzimidazolylidenes failed completely (entries 1 and 2), thiazolylidenes derived from 3 and 49 resulted in the formation of small amounts of product (entries 3 and 4). This could be somewhat improved by the use of the thiazolylidene derived from 5, commonly applied in NHC-organocatalysis5 (entry 5). Interestingly, however, the major improvement came with employing the NHC derived from 6 (entry 6), which was designed by us for dual organo-/metal-catalysis.11 Further optimization of base and solvent proved N(iPr)2Et in THF to be the best combination (entry 8), providing the hydroxymethylated product in 74% yield (based on 1H NMR).
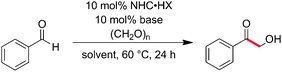
| Entry | NHC·HX | Base | Solvent | Yielda (%) |
|---|---|---|---|---|
a Yield determined by 1H NMR (mesitylene as internal standard).
b 2 eq. (CH2O)n, CPhCHO = 0.5 mol L−1.
c 1 eq. (CH2O)n, CPhCHO = 0.5 mol L−1.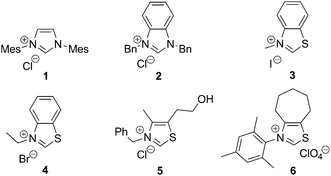
|
||||
| 1b | 1 | NEt3 | THF | 0 |
| 2b | 2 | NEt3 | THF | 0 |
| 3b | 3 | NEt3 | THF | 9 |
| 4b | 4 | NEt3 | THF | 15 |
| 5b | 5 | NEt3 | THF | 26 |
| 6b | 6 | NEt3 | THF | 66 |
| 7c | 6 | NEt3 | THF | 53 |
| 8c | 6 | N(iPr)2Et | THF | 74 |
| 9c | 6 | DBU | THF | 53 |
| 10c | 6 | N(iPr)2Et | t AmylOH | 66 |
| 11c | 6 | N(iPr)2Et | DMF | 68 |
| 12c | 6 | N(iPr)2Et | Toluene | 57 |
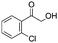
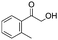
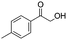
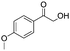
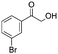
After additional optimization,12 we investigated the scope of this potentially useful hydroxymethylation reaction, employing 3 eq. of paraformaldehyde (Table 2).13 Several different classes of aldehydes proved suitable. First, the hydroxymethylation of benzaldehyde provided 70% of the desired product, on a 1 mmol as well as on a 10 mmol scale (entry 1).§ However, generally, 1H NMR analysis of the crude reaction mixture indicates a significantly higher yield. Some of the product might be lost upon column chromatography. This notion was validated by a simple control experiment: reduction of the crude reaction product with NaBH4 resulted in the formation of the corresponding diol product, which could be isolated in an improved yield of 79%.13 Several para-substituted benzaldehyde derivatives were smoothly hydroxymethylated in good yields, with electron-poor aldehydes leading to higher yields than electron-rich ones (entries 2–6). Although o-chloro benzaldehyde reacts well (entry 8), ortho-substitution results in reduced yields in some cases (entries 9 and 10). This kind of deterioration is often observed in NHC-catalyzed umpolung chemistry due to the increased steric shielding of the aldehyde moiety of these substrates. Several halide-containing hydroxymethyl ketones can readily be formed (entries 2, 3, 7 and 8), allowing further functionalization by standard cross-coupling methodology. The use of o-propargyloxy-benzaldehyde (entry 10) also represents an interesting competition experiment, since this substrate is known to undergo an NHC-catalyzed hydroacylation of the alkyne moiety.7d Under standard conditions, however, only the desired hydroxymethylation product was observed.

| Entry | Product | Yielda (%) | Entry | Product | Yielda (%) |
|---|---|---|---|---|---|
| a Isolated yield after column chromatography. Reactions were run on a 1 mmol scale. b Yield determined after 6 h. c On a 10 mmol scale, a similar yield of 70% was obtained. d Yield determined after 20 h. e Yield determined by 1H NMR; reaction on 0.25 mmol scale. f Yield determined after 12 h. | |||||
| 1 |
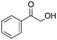
|
70b,c | 8 |
|
77e |
| 2 |
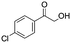
|
77d | 9 |
|
33 |
| 3 |

|
74 | 10 |
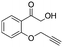
|
50 |
| 4 |
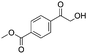
|
86 | 11 |
|
57 |
| 5 |
|
60 | 12 |
|
64f |
| 6 |
|
32 | 13 |
|
57e |
| 7 |
|
85c | 14 |
|
29 |
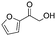
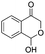
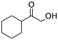
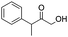
Moreover, the smooth hydroxymethylation of 2-furylcarbaldehyde and the mono-hydroxymethylation/cyclization of benzene-1,2-dicarbaldehyde are remarkable (entries 11 and 12). The benzopyranone, formed in the latter reaction in 64% yield, is difficult to obtain with alternative methods.14 In addition, non-aromatic aldehydes can also be hydroxymethylated (entries 13 and 14).
A straightforward mechanistic scenario is shown in Scheme 1. The sequence involves the nucleophilic attack of the NHC onto the aldehyde group (step 1), proton transfer giving the Breslow intermediates A or B (step 2), and finally the addition to an electrophilic aldehyde group, followed by proton transfer and elimination of the NHC catalyst (step 3). Under the optimized reaction conditions and utilizing thiazolium salt 6 as the NHC precursor, 7 and 9 are formed, with 7 being the predominant product. In addition, small amounts of 8 could be found, whereas 10 was not detectable at all.15 Considering the high electrophilicity and steric accessibility of CH2O, it is reasonable that the Breslow intermediates A and B preferentially attack formaldehyde, resulting in the observed product distribution.
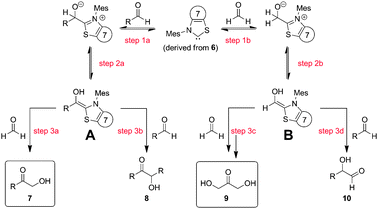 | ||
| Scheme 1 Plausible mechanistic pathways. | ||
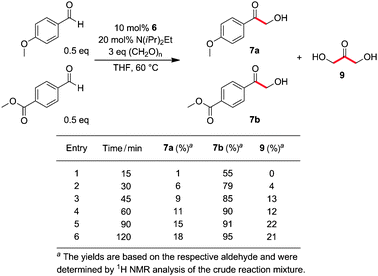
In order to shed some light on the mechanism, the following experiments were run. First, the reversibility of the formation of benzoin 8 under hydroxymethylation conditions was demonstrated by employing 8 (R = Ph) as a substrate, resulting in the formation of 27% of the corresponding product 7.13 Second, on the contrary, hydroxymethylation product 7 (R = Ph) was found to be stable under reaction conditions.13,16 Finally, a simple competition experiment was insightful (Scheme 2). Treatment of an electron-rich and an electron-poor benzaldehyde derivative in the same flask under standard conditions strikingly showed the superior reactivity of more electrophilic aldehydes, resulting in the rapid formation of hydroxymethyl ketone 7b. 7a and dihydroxyacetone (9) are formed at approximately the same rate but significantly slower than 7b. This seems to indicate that the addition of the Breslow intermediate to formaldehyde is not the rate-determining step of this transformation. Finally, it should be noted that a low equilibrium concentration of free CH2O might also play a role for the observed product distribution.
 | ||
| Scheme 2 Competition experiment. | ||
In conclusion, we have developed a direct NHC-catalyzed hydroxymethylation of aldehydes that tolerates several important functional groups. In addition, some light is shed on the mechanistic intricacies of this remarkably selective transformation. The rather broad scope and the attractive features of this transformation should result in ample application of this method.
Financial support by the Deutsche Forschungsgemeinschaft (SPP 1179) is gratefully acknowledged. The research of F.G. was supported by the Alfried Krupp Prize for Young University Teachers of the Alfried Krupp von Bohlen und Halbach Foundation. We also thank Dr A. T. Biju and I. Piel for helpful discussions.
Notes and references
- (a) D. Villemin, N. Cheikh, B. Mostefa-Kara, N. Bar, N. Choukchou-Braham and M. A. Didi, Tetrahedron Lett., 2006, 47, 5519 CrossRef CAS; (b) E. Lipka, M. P. Vaccher, C. Vaccher and C. Len, Bioorg. Med. Chem. Lett., 2005, 15, 501 CrossRef CAS; (c) C. Len, A. Selouane, A. Weiling, F. Coicou and D. Postel, Tetrahedron Lett., 2003, 44, 663 CrossRef CAS; (d) F. Babudri, V. Fiandanese, G. Marchese and A. Punzi, Tetrahedron, 1999, 55, 2431 CrossRef CAS.
- F. F. Wong, P.-W. Chang, H.-C. Lin, B.-J. You, J.-J. Huang and S.-K. Lin, J. Organomet. Chem., 2009, 694, 3452 CrossRef CAS.
- (a) C. Chen, X. Feng, G. Zhang, Q. Zhao and G. Huang, Synthesis, 2008, 3205; (b) Y. Zhang, Z. Shen, J. Tang, Y. Zhang, L. Kong and Y. Zhang, Org. Biomol. Chem., 2006, 4, 1478 RSC; (c) B. Plietker, Eur. J. Org. Chem., 2005, 1919 CrossRef CAS; (d) B. Plietker, J. Org. Chem., 2004, 69, 8287 CrossRef CAS; (e) B. Plietker, J. Org. Chem., 2003, 68, 7123 CrossRef CAS; (f) A. K. El-Qisairi and H. A. Qaseer, J. Organomet. Chem., 2002, 659, 50 CrossRef CAS; (g) F. A. Davis and A. C. Sheppard, J. Org. Chem., 1987, 52, 954 CrossRef CAS; (h) E. Vedejs, J. Am. Chem. Soc., 1974, 96, 5944 CrossRef CAS.
- For authoritative reviews on NHCs and their application as ligands in transition-metal catalysis, see: (a) A. J. Arduengo III., Acc. Chem. Res., 1999, 32, 913 CrossRef; (b) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef; (c) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (d) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CAS; (e) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS; (f) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (g) N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin, 2007 Search PubMed; (h) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CrossRef CAS; (i) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS; (j) S. Würtz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523 CrossRef CAS; (k) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef CAS; (l) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 CrossRef.
- For excellent reviews, see: (a) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS; (c) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS; (d) E. M. Phillips, A. Chan and K. A. Scheidt, Aldrichimica Acta, 2009, 42, 55 CAS.
- For selected examples of Benzoin reactions, see: (a) D. Enders and U. Kallfass, Angew. Chem., Int. Ed., 2002, 41, 1743 CrossRef CAS; (b) D. Enders, O. Niemeier and T. Balensiefer, Angew. Chem., Int. Ed., 2006, 45, 1463 CrossRef CAS; (c) H. Takikawa, Y. Hachisu, J. W. Bode and K. Suzuki, Angew. Chem., Int. Ed., 2006, 45, 3492 CrossRef CAS; (d) M. J. White and F. J. Leeper, J. Org. Chem., 2001, 66, 5124 CrossRef CAS; (e) D. Enders and A. Henseler, Adv. Synth. Catal., 2009, 351, 1749 CrossRef CAS.
- For reviews on the Stetter reaction, see: (a) J. Read de Alaniz and T. Rovis, Synlett, 2009, 1189; (b) M. Christmann, Angew. Chem., Int. Ed., 2005, 44, 2632 CrossRef CAS. For addition of Breslow intermediates to non-activated alkenes and alkynes, see: (c) K. Hirano, A. T. Biju, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 14190 CrossRef CAS; (d) A. T. Biju, N. E. Wurz and F. Glorius, J. Am. Chem. Soc., 2010, 132, 5970 CrossRef CAS; (e) J. He, S. Tang, J. Liu, Y. Su, X. Pan and X. She, Tetrahedron, 2008, 64, 8797 CrossRef CAS. See also: (f) J. Read de Alaniz, M. S. Kerr, J. L. Moore and T. Rovis, J. Org. Chem., 2008, 73, 2033 CrossRef CAS; (g) D. Enders, K. Breuer and J. Runsink, Helv. Chim. Acta, 1996, 79, 1899 CAS.
- For selected work in homoenolate chemistry, see: (a) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 CrossRef CAS; (b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS; (c) M. He and J. W. Bode, Org. Lett., 2005, 7, 3131 CrossRef CAS; (d) V. Nair, S. Vellalath, M. Poonoth and E. Suresh, J. Am. Chem. Soc., 2006, 128, 8736 CrossRef CAS; (e) C. Burstein, S. Tschan, X. Xie and F. Glorius, Synthesis, 2006, 2418 CrossRef CAS; (f) A. Chan and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 5334 CrossRef CAS; (g) E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2008, 130, 2416 CrossRef CAS.
- T. Matsumoto, M. Ohishi and S. Inoue, J. Org. Chem., 1985, 50, 603 CrossRef CAS.
- (a) A. S. Demir, P. Ayhan, A. C. Igdir and A. N. Duygu, Tetrahedron, 2004, 60, 6509 CrossRef CAS; (b) A. Cosp, C. Dresen, M. Pohl, L. Walter, C. Röhr and M. Müller, Adv. Synth. Catal., 2008, 350, 759 CrossRef CAS.
- (a) R. Lebeuf, K. Hirano and F. Glorius, Org. Lett., 2008, 10, 4243 CrossRef CAS. See also: (b) K. Hirano, I. Piel and F. Glorius, Adv. Synth. Catal., 2008, 350, 984 CrossRef CAS.
- Further screening of different amounts of catalyst, paraformaldehyde and base, showed 10 mol% of the catalyst precursor 6, 3 eq. of the paraformaldehyde and a ratio of base (N(iPr)2Et) to salt 6 of 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 to be optimal.
∶1 to be optimal. - See ESI‡ for further details.
- A. R. Butler and I. Hussain, J. Chem. Soc., Perkin Trans. 2, 1981, 310 RSC.
- At this point, the intermediate formation of 10 and its conversion to the observed product 7 by keto–enol tautomerism cannot be excluded. However, whereas the acid catalyzed conversion of hydroxy phenyl acetaldehyde to hydroxymethyl phenyl ketone is reported, to the best of our knowledge the base catalyzed one is not ( (a) F. Weygand, H. J. Bestmann, H. Ziemann and E. Klieger, Chem. Ber., 1958, 91, 1043 CrossRef CAS). For literature reports on the formation of hydroxy phenyl acetaldehyde, see: ( (b) D. Bojer, I. Kamps, X. Tian, A. Hepp, T. Pape, R. Fröhlich and N. W. Mitzel, Angew. Chem., Int. Ed., 2007, 46, 4176 CrossRef CAS; (c) P. Tinapp, Chem. Ber., 1971, 104, 2266 CrossRef CAS ).
- However, (ir)reversibility seems to depend on the electronic nature of the R substituent: whereas 7 (R = Ph) was stable under reaction conditions, the compound 7 substituted with an electron-withdrawing ester group (R = 4-CO2Me–C6H4) was not (ref. 13).
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization data. See DOI: 10.1039/c0cc02416c |
| § General hydroxymethylation procedure: in an oven-dried Schlenk-flask sealed with a rubber septum, thiazolium salt 6 (37 mg, 0.1 mmol, 0.1 eq.), paraformaldehyde (90 mg, 3.0 mmol, 3.0 eq.) and the aldehyde (1.0 mmol, 1.0 eq.) were suspended in dry THF (4 mL). N(iPr)2Et (33 μL, 0.2 mmol, 0.2 eq.) was added and the resulting mixture was heated to 60 °C. In the case of aliphatic aldehydes, the aldehyde was added after stirring the other reactants for 5 min at room temperature. After a maximum reaction time of 24 h, the solvent was evaporated and the crude product pre-adsorbed on Celite. Flash chromatography on silica gel afforded the pure product. |
| This journal is © The Royal Society of Chemistry 2011 |