Rational design of single-molecule magnets: a supramolecular approach†
Thorsten
Glaser
*
Lehrstuhl für Anorganische Chemie I, Fakultät für Chemie, Universität Bielefeld, Universitätsstr. 25, D-33615 Bielefeld, Germany. E-mail: thorsten.glaser@uni-bielefeld.de; Fax: +49 521-106-6003
First published on 22nd September 2010
Abstract
Since the discovery that Mn121212OAc acts as a single-molecule magnet (SMM), an increasing number of transition metal complexes have been demonstrated to behave as SMMs. The signature of a SMM is a slow relaxation of the magnetization at low temperatures accompanied by a magnetic hysteresis. The origin of SMM behaviour is the existence of an appreciable thermal barrier U for spin-reversal called magnetic anisotropy barrier which is related to the combination of a large total spin ground state (St) and an easy-axis magnetic anisotropy. The extensive research on Mn121212OAc and other SMMs has established more prerequisites for a rational development of new SMMs besides the high-spin ground state and the magnetic anisotropy: the symmetry should be at least C3 to minimize the quantum tunneling of the magnetization through the anisotropy barrier but lower than cubic to avoid the cancellation of the local anisotropies upon projection onto the spin ground state. Based on these prerequisites, we have designed the ligand triplesalen which combines the phloroglucinol bridging unit for high spin ground states by the spin-polarization mechanism with a salen-like ligand environment for single-site magnetic anisotropies by a strong tetragonal ligand field. The C3 symmetric, trinuclear complexes of the triplesalen ligand (talent-Bu2)6− exhibit a strong ligand folding resulting in an overall bowl-shaped molecular structure. This ligand folding preorganizes the axial coordination sites of the metal salen subunits for the complementary binding of three facial nitrogen atoms of a hexacyanometallate unit. This leads to a high driving force for the formation of heptanuclear complexes [Mttt66Mccc]n+nn+ by the assembly of three molecular building blocks. Attractive van der Waals interactions of the tert-butyl phenyl units of two triplesalen trinuclear building blocks increase the driving force. In this respect, we have been able to synthesize the isostructural series [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+, [MnIIIIII66FeIIIIII]3+3+, and [MnIIIIII66CoIIIIII]3+3+ with [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ being a SMM. A detailed analysis and comparison of the magnetic properties of the three heptanuclear complexes and the tetranuclear half-unit [MnIIIIII33CrIIIIII]3+3+ provides significant insight for further optimization of the SMM properties. The modular assembly of the heptanuclear complexes from three molecular building blocks allows the fine-tuning of the molecular and steric properties of each building block without losing the driving force for the formation of the heptanuclear complexes. This possibility of rational improvements of our isostructural series is the main advantage of our supramolecular approach.
 Thorsten Glaser | Thorsten Glaser studied chemistry in Bochum and obtained his Dr. rer. nat. in 1997 with Prof. K. Wieghardt at the Max-Planck-Institut, Mülheim. After postdoctoral work with Professors E. I. Solomon and K. O. Hodgson at Stanford University, he started his independent research in 2000 in Münster with a Liebig fellowship of the Fonds of the Chemical Industry and obtained the venia legendi in 2004. He was awarded the ADUC prize in 2002, the Werner-von-Siemens-Medal in 2004, the Young-Talent-Award of Münster University in 2004, and the “Goldener Brendel” from the students for the best chemistry lecture. In 2005 he declined an offer for a full professorship at Marburg University and accepted an offer for a full professorship at Bielefeld University. Since 2008 he is speaker of the DFG research unit “Nanomagnets”. |
1. Introduction
The broad field of molecular magnetism is concerned with the understanding of the magnetic properties of molecules exhibiting one or more magnetic centers in form of transition metal ions or organic radicals.1–3 Besides the demand to understand the magnetic properties of a single magnetic center (especially to correlate anisotropic properties to the molecular structure) the study and interpretation of intramolecular and intermolecular interactions between magnetic centers are the main focus of molecular magnetism. In the last two decades, the synthesis and characterization of molecule-based magnetic materials, i.e. compounds with the characteristic features of conventional solid-state magnets but prepared with synthetic tools of molecular chemistry under kinetic control, developed to a contemporary application of molecular magnetism.4–8 The considerable interest in the synthesis of magnetic materials based on molecular entities with its potential applications in fields like molecular electronics9,10 started with the observation that [Cp*2Fe]+[TCNE]− exhibits a spontaneous long-range ferromagnetic ordering with a Curie temperature TC of 4.8 K.11,12 This 3D long-range magnetic order in a system constructed from molecular building blocks initiated world-wide research activities to prepare quasi-1D, 2D, and 3D molecule-based magnets. Prominent examples with TC's above room temperature are V(TCNE)2·y(CH2Cl2)13 and some members of the family of Prussian Blue analogues like KVII[CrIII(CN)6]14 or VII0.42VIII0.58[CrIII(CN)6]0.86·2.8H2O.15,16 This last class based on the use of hexacyanometallates [M(CN)6]n− provides a convenient approach for the design of new interesting materials because the high symmetry of the M–CN–M′ exchange pathway allows the prediction of the nature of the exchange interactions17–20 by the use of the Goodenough–Kanamori rules.21–23Another milestone in the field of molecule-based magnetic materials was the observation that [Mn12O12(OAc)16(OH2)4], Mn121212OAc (Fig. 1a and b),24 exhibits a hysteresis in the magnetization of pure molecular origin25,26 and has been the first member of a new class of molecule-based magnets called single-molecule magnets27 (SMMs).28–31 These finite-size molecules possess an energy-barrier U for spin reversal causing a slow relaxation of the magnetization at low temperatures. This magnetic bistability of SMMs promises access to devices for ultimate high-density memory storage and quantum computing.29,32,33 Since the report that Mn121212OAc exhibits SMM behaviour, a still increasing number of SMMs have been identified in the effort to enhance the magnetic anisotropy barrier.34,35
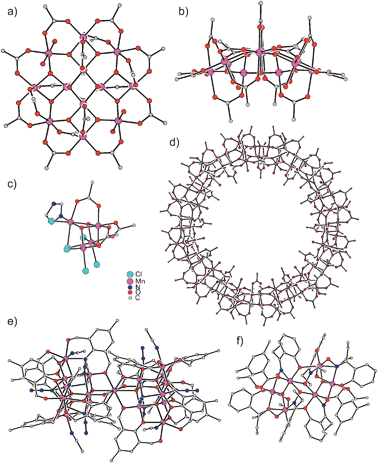 | ||
| Fig. 1 Molecular structures of (a) and (b) Mn121212OAc,24 (c) Mn44,27 (d) Mn8484,49 (e) Mn1919,50 (f) Mn66.56 | ||
This article presents a personal account of work in the author's research group over the last years, in which the essence of previous investigations on SMMs has been used as a basis for a rational ligand and complex design of a new family of SMMs based on the ligand triplesalen.
2. Design principles
2.1 The properties of Mn121212OAc as a guide
Mn121212OAc consists of 8 MnIII and 4 MnIV ions which couple by exchange interactions to a total spin ground state St = 10 of the whole molecule.26 An important property of this spin is its anisotropy, i.e. it has a preferred orientation in the molecular framework that is perpendicular to the approximate molecular plane (Fig. 1a). As a pure spin is magnetically completely isotropic, the anisotropy arises from some orbital angular momentum contribution. This magnetic anisotropy can be phenomenologically described by the so-called zero-field splitting, which is parameterized by an axial zero-field splitting parameter D and a rhombicity E/D. A spin of quantum number S possesses 2S + 1 sublevels of quantum number MS. In zero magnetic field, these sublevels are degenerate. The effect of an axial zero-field splitting (Ĥ = DŜz2 − ⅓(S(S + 1))) is schematically shown in Fig. 2a for an St = 10 spin with negative D. The MS sublevels split with MS = ±S lying energetically lowest. This corresponds to the preferred orientation of the spin perpendicular to the molecular plane, while other orientations correspond to the MS states of lower absolute value, which lie energetically higher. This is also called an easy-axis magnetic anisotropy. A positive D corresponds to a preferred orientation of the spin in the molecular plane associated with the MS = 0 (or ±1/2 for non-integer spins) being lowest in energy. This is referred to as an easy-plane anisotropy.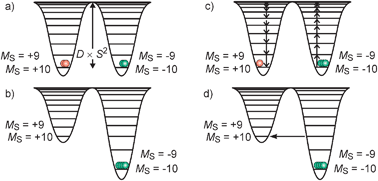 | ||
| Fig. 2 Schematic representation of the anisotropy barrier in SMMs: (a) effect of a negative zero-field splitting parameter on a St = 10 spin manifold; (b) magnetization of the sample by application of an external magnetic field (Zeeman effect); (c) frozen magnetized sample showing a slow relaxation of the magnetization over the anisotropy barrier after turning off the external magnetic field; (d) quantum tunneling of the magnetization through the anisotropy barrier for magnetic fields leading to interacting MS substates at the same energy. Note that the representation as potentials is only a guide to the eyes. The basic representation is energy as a function of the MS quantum number. | ||
The effect of the application of an external magnetic field is illustrated in Fig. 2b. The states with negative MS values become energetically stabilized while the substates with positive MS values become energetically destabilized (Zeeman effect). This leads to a preferred population of the MS = −10 substate corresponding to a preferred orientation of the microscopic magnetic moments ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) in the direction of the external magnetic field. The effect is a magnetization of the macroscopic sample. This phenomenon occurs in any paramagnetic sample. By switching off the external magnetic field the unperturbed energies (Fig. 2a) are restored. While a paramagnetic system relaxes very fast (≪1 s, dependent on temperature)36 to the unmagnetized equilibrium initial state, this relaxation of the magnetization is slow in a SMM due to the anisotropy barrier schematized in Fig. 2c. This energy barrier (DSt·St2 for integer spins, DSt·(St2 − 1/4) for non-integer spins) for spin reversal originates from a ground state with large total spin St and large magnetic anisotropy of the easy-axis type. In Mn121212OAc, the St = 10 spin ground state has a zero-field splitting DSt=10 ≈ −0.5 cm−1, which results in an anisotropy barrier of ∼50 cm−1.25,30 In simple words, the microscopic magnetic moments are frozen in a non-randomized assembly so that a macroscopic magnetization remains without an external magnetic field. Thermally, the direction of the magnetization can only be reversed by a pathway over the top of the energy barrier.31 In this respect, the anisotropy barrier represents an activation energy for the magnetization reversal. Below some temperature, the relaxation is slow enough to be probed by experimental techniques with regard to the specific time scales of the techniques.
in the direction of the external magnetic field. The effect is a magnetization of the macroscopic sample. This phenomenon occurs in any paramagnetic sample. By switching off the external magnetic field the unperturbed energies (Fig. 2a) are restored. While a paramagnetic system relaxes very fast (≪1 s, dependent on temperature)36 to the unmagnetized equilibrium initial state, this relaxation of the magnetization is slow in a SMM due to the anisotropy barrier schematized in Fig. 2c. This energy barrier (DSt·St2 for integer spins, DSt·(St2 − 1/4) for non-integer spins) for spin reversal originates from a ground state with large total spin St and large magnetic anisotropy of the easy-axis type. In Mn121212OAc, the St = 10 spin ground state has a zero-field splitting DSt=10 ≈ −0.5 cm−1, which results in an anisotropy barrier of ∼50 cm−1.25,30 In simple words, the microscopic magnetic moments are frozen in a non-randomized assembly so that a macroscopic magnetization remains without an external magnetic field. Thermally, the direction of the magnetization can only be reversed by a pathway over the top of the energy barrier.31 In this respect, the anisotropy barrier represents an activation energy for the magnetization reversal. Below some temperature, the relaxation is slow enough to be probed by experimental techniques with regard to the specific time scales of the techniques.
2.2 Recent highlights past Mn121212OAc
Since the discovery of the fascinating SMM behavior of Mn121212OAc, a lot of synthetic efforts have been devoted to the preparation of new molecules with an increased anisotropy barrier and fascinating new structural motives have been reported. Besides the serendipitous approaches,37 the majority of work is based on synthetic approaches to increase the anisotropy barrier by increasing the total spin ground state St while providing some source of magnetic anisotropy.29,34,35,38–40 Here, only some very selective examples can be presented.2.3 High spin ground states
The height of the anisotropy barrier U clearly defines two necessary requirements to be considered for the development of new SMMs: a high spin ground state St combined with a strong magnetic anisotropy DSt of the easy-axis type. Unfortunately, an important result from the study of exchange interactions between paramagnetic metal ions transmitted through bridging ligands (superexchange) is that the predominant coupling is of antiferromagnetic nature. This observation is well understood on the basis of several levels of theory and is mainly attributed to the overlap of magnetic orbitals (orbitals containing an unpaired electron) (1st Goodenough–Kanamori rule)21–23 and the onset of chemical bonding, i.e. the pairing of electrons in the resulting bonding molecular orbitals.1Thus, the need for high spin ground states St to rationally develop new SMMs requires synthetic strategies to building blocks, which either enforce ferromagnetic interactions and/or predictable antiferromagnetic couplings resulting in ferrimagnetic arrangements of the spins. This objective was already formulated by the late Olivier Kahn: ‘The normal trend for the molecular state is the pairing of electrons […] with a cancellation of the electron spins. The design of a molecule-based magnet requires that this trend be successfully opposed’.60
In order to establish synthetic recipes for parallel spin alignments we are exploring three different strategies: (1) the double exchange mechanism61–63 in face-sharing octahedra,64–67 (2) the use of orthogonal magnetic orbitals (2nd Goodenough–Kanamori rule),21–23,68,69 and (3) the spin-polarization mechanism70–74 in m-phenylene-bridged complexes, which is the strategy employed in this project. We have recently summarized the principle aspects and the application of the spin-polarization mechanism75 so that only the main concept of this simple, heuristic phenomenon will be recapitulated here.
It is well established in organic chemistry76,77 that the m-phenylene linkage of organic radicals and carbenes leads to ferromagnetic interactions, while in the corresponding o- and p-phenylene linkages the interaction is antiferromagnetic (Scheme 1, left). Application of many-electron MO theory provides an alternation of the sign of the spin-density at the atoms in the bridging unit.78 This is illustrated in Scheme 1, right and provides the opportunity to develop high spin organic radicals and carbenes on the back of an envelope as the nature of the interaction – ferromagnetic or antiferromagnetic – only depends on the number of atoms between the paramagnetic centers.79
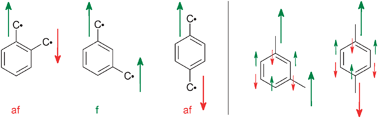 | ||
| Scheme 1 | ||
This concept has been applied to transition metal complexes by using various types of bridging ligands based on heteroaromatic systems,80–83trithiocyanuric acid,84resorcinol,85,86 and di- and trioxamato benzene systems.87–90 One trinuclear complex using 1,3,5-trihydroxybenzene (phloroglucinol) as a bridging ligand was reported which exhibits ferromagnetic interactions between three (Tp*)MoVOCl fragments.91 Our approach was to use phloroglucinol to establish ferromagnetic interactions between paramagnetic 3d ions, as their local spin can be up to Si = 5/2, which is difficult to achieve with 4d or 5d metal ions due to their intrinsic preference for low-spin electronic configurations. However, we realized by painful failures in a whole series of synthetic approaches that the preparation of trinuclear complexes of 3d metal ions bridged by the parent phloroglucinol is hindered due to too low stabilities. To overcome this problem we have attached pendant arms with additional coordinating groups in the 2,4,6-positions of phloroglucinol, thereby enhancing the stability by the chelate effect. In this respect, the use of the tris-imine ligand H3L (Scheme 2a) allowed the preparation of the trinuclear complexes [L(CuII(bpy))3](BF4)3 and [L(CuII(sal))3] (Hsal = salicylaldehyde), which exhibit ferromagnetic couplings with J = +1.96 cm−1 and J = +3.31 cm−1, respectively (all J values are given according to the HDvV-Hamiltonian in the form H = −2 JS1S2).92 Thus, the phloroglucinol bridging unit propagates ferromagnetic couplings in trinuclear CuII complexes, while the magnitude of the coupling is relatively small.
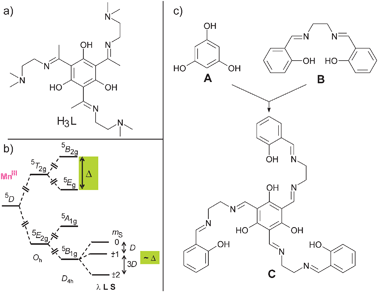 | ||
| Scheme 2 | ||
2.4 Magnetic anisotropy
The main challenge in the preparation of SMMs with increased anisotropy barriers is the realization of both a high spin ground state St and a sizable magnetic anisotropy DSt in one molecule. The magnetic anisotropy of the ground state (DSt) of polynuclear transition metal complexes has its main contribution usually from the projection of the single-site anisotropies (Di) onto the spin ground state St. Dipolar and anisotropic interactions usually yield only minor contributions.93–95 Thus, the local magnetic moments should possess some source of anisotropy. The magnetic moment arising from a pure spin angular momentum is not coupled to the molecular framework and exhibits no magnetic anisotropy. The magnetic anisotropy results from a certain amount of orbital angular momentum in the magnetic moment. Transition metal ions with a formal T ground state possess an in-state first-order orbital angular momentum,96 so that the application of the pure spin-Hamiltonian is not indicated. However, this first-order orbital angular momentum gives rise to very strong magnetic anisotropies. Transition metal ions with formal A, B, or E ground states do not possess a first-order orbital angular momentum but some orbital angular momentum may arise from the mixing of the excited states into the ground state by second-order spin–orbit coupling. This leads to the anisotropy of the spin ground state which is parameterized in the spin-Hamiltonian by the zero-field splitting D.97Spin–orbit coupling is a relativistic effect and scales with Z4. Thus, 4d and 5d metal ions exhibit stronger magnetic anisotropies than 3d metal ions. However, 4d and 5d metal ions are, besides some spectacular exceptions, in a low-spin configuration with small spin quantum numbers Si. Lanthanide and actinide ions may also possess strong magnetic anisotropies. There is an increasing number of SMMs containing lanthanide ions98–102 and there are even mononuclear complexes of therbium, which exhibit a slow relaxation of the magnetization as SMMs.103–105 The drawback of the use of lanthanide ions is that they exhibit only weak exchange couplings due to the buried nature of their magnetic f orbitals and thus their low covalencies.
In summary, the main problem in a rational design of SMMs is the lack of metal ions, which combine strong magnetic anisotropies with high local spin states and strong exchange couplings. In this respect, the use of 3d metal ions is not a natural choice but a compromise between high local spin states combined with reasonably strong exchange couplings, while the magnetic anisotropies are usually small.
As magnetic anisotropy is a complicated property, the perturbational treatment in ligand-field theory is educational and valuable for the synthetic coordination chemist to obtain some recipes for introducing magnetic anisotropy. The origin of the magnetic anisotropy of MnIII ions, which is the main source of anisotropy in Mn121212OAc, should illustrate how the careful choice of the ligand environment may provide access to appreciable magnetic anisotropy even for 3d ions. MnIII is a d4 ion and the free ion has a 5D ground state. This state is split in an octahedral ligand field (Oh) into a 5E2g ground state and a 5T2g excited state (Scheme 2b). The 5E2g ground state in pure Oh symmetry is magnetically completely isotropic. The well-known magnetic anisotropy of MnIII ions can be explained by ligand-field theory through the incorporation of two perturbations.106–108 The 5E2g ground state in Oh symmetry is Jahn–Teller active and MnIII complexes usually exhibit a tetragonal elongation, which results in a symmetry reduction to D4h. This leads to a 5B1g ground state (Scheme 2b), which is still magnetically completely isotropic. Spin–orbit coupling mixes the excited 5E2g and 5B2g states into the ground state, which results in a splitting of the 5B1g ground state. This splitting is phenomenologically described by the zero-field splitting (Scheme 2b). Second-order perturbation theory provides that this splitting is proportional to the splitting Δ of the 5T2g state induced only by the symmetry reduction from Oh to D4h.107,109 Thus, the stronger the tetragonal symmetry reduction, the stronger the splitting of the 5B1g state and the zero-field splitting. Therefore, the use of a ligand, which introduces a strong tetragonal component into the ligand field, should maximize the magnetic anisotropy.
Besides the careful consideration of the local magnetic anisotropies, there are also some symmetry constraints for the relative orientation of the metal ions. Since zero-field splittings are tensor quantities, the projection of the single-site zero-field splittings onto the spin ground state may vanish as the metal ion arrangement approaches a cubic symmetry.93
2.5 Quantum tunnelling and its consequences
The fascinating properties and the still ongoing interest in the study of Mn121212OAc are not only due to the slow relaxation of the magnetization, but also to the presence of quantum effects.110–113 While the height of the anisotropy barrier U is related to the thermal pathway for spin reversal, there is also a non-thermal pathway for spin reversal through the anisotropy barrier by a quantum mechanical tunnelling of the magnetization (Fig. 1d). This tunnelling mechanism is directly related to the rhombic component (Ĥ = E(Ŝx2 − Ŝy2))30 of the magnetic anisotropy which vanishes for complexes with at least a threefold axis (see Sections 2.2.1 and 2.2.2). This provides another prerequisite to improve the properties of SMMs by the minimization of the quantum mechanical magnetization tunneling, which provides an alternative pathway for spin-reversal and thus the loss of information.2.6 Combination into a hybrid ligand
In summary, a rational design of a SMM requires not only a high spin ground state combined with a significant magnetic anisotropy of the easy axis type, but should also imply considerations of the topology of the metal ion arrangement. The overall molecular symmetry should be at least trigonal to minimize the quantum tunneling but lower than cubic to avoid an isotropic system. Such a control is difficult to achieve with small flexible ligands of low directionality. The use of the concepts of supramolecular chemistry (i.e. association of molecular building blocks based on (i) preorganization and topological constraints to increase binding power and (ii) complementarity to determine molecular recognition)114–116 should provide an ansatz for a rational design of SMMs. An advantage of this modular approach is that the molecular building blocks can be fine-tuned by steric and electronic controls while the driving forces to assemble the whole chemical entity are still present (see, e.g., the variation from Mn1919 to Mn1818Dy in Section 2.2.4). It is important to point out that rational design should not be misinterpreted such that structural parameters (bond distances, angles, torsion angles) or the extent of Jahn–Teller distortions can be exactly predetermined or changed into values, which would result in better magnetic properties.In this respect, we have tried a ligand and complex design based on all these requirements for SMMs. We thought to combine the phloroglucinol bridging unit A with the coordination environment of a salen ligand B into the C3 symmetric ligand triplesalen C (Scheme 2c). The design of the triplesalen ligand has been based on the following concepts: in order to ensure high spin ground states, we have chosen the phloroglucinol bridging unit A, which is able to transmit ferromagnetic interactions in trinuclear CuII complexes by the spin-polarization mechanism (see Section 2.3). We have chosen a salen-like coordination environment because the salen ligand B is known to establish pronounced magnetic anisotropies by its strong ligand field in the basal plane (see Section 2.4).94,106,108 A well studied example is the Jacobsen catalyst [(salen′)MnIIICl] (H2salen′ = (R,R)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamine) which is a MnIII (S = 2) species with a zero-field splitting of D ≈ −2.5 cm−1.108,117,118 In this respect, it is interesting to note that already dimeric MnIII salen complexes behave as SMMs.119–121 Furthermore, the trigonal symmetry provided by the triplesalen ligand should result in highly axial anisotropies with small to zero rhombic components and thus reduced tunneling through the anisotropy barrier (see Section 2.5).
3. Ligand synthesis
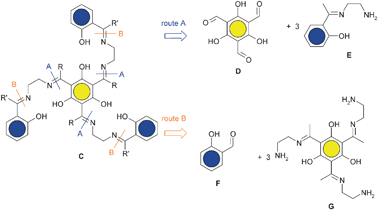
Symmetrical salen ligands are very common and can easily be prepared by one pot condensation reactions of one diamine with two identical aldehyde or ketone derivatives. However, the targeted triplesalen ligand C contains three unsymmetrically substituted ethylene diamine units making the synthesis not trivial. The controlled synthesis of unsymmetrical salen ligands (not a mixture of symmetrical and unsymmetrical compounds) containing two different aldehyde or ketone derivatives must involve in a first step the isolation of a mono-condensated ethylene diamine, a so-called salen half-unit.122–126 This intermediate would provide the opportunity for the synthesis of the desired unsymmetrical salen ligand by the reaction of the remaining amine with the other aldehyde or ketone derivative. However, a salen half-unit is not always accessible123 and mainly requires a differentiation of the reactivity of the two amine functions.127 Another important aspect to consider is that an aldimine is usually easy to prepare but not very stable. On the other hand, the preparation of a ketimine requires harsher conditions but it exhibits a higher stability.128 Therefore, in a stepwise procedure, the generation of the more stable ketimine half-unit in the first step and the subsequent reaction with an aldehyde is a promising sequence.Two reaction routes are in principle feasible for the preparation of the targeted triplesalen ligand C (Scheme 3). Route A requires the synthesis of a ketimine salen half-unit E in the first step which reacts in the second step with the trisaldehyde derivative of phloroglucinol D. Route B requires the synthesis of a triplesalen ketimine half-unit G which reacts in the second step with a salicylaldehyde derivative F.
 | ||
| Scheme 3 | ||
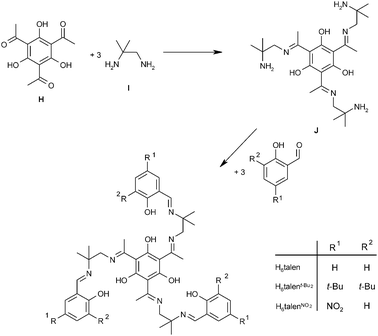
Thus, the key step in the synthesis of a triplesalen ligand C is the preparation of a half-unit, either E or G. Several methods for the preparation and isolation of derivatives of the parent salen half-unit E are described in the literature.124,125,127,129,130 We applied several of these methods for the synthesis of a triplesalen ligand and obtained various new compounds.131 The successful synthesis of a triplesalen ligand is based on an observation of Elias and coworkers.127 They reported that the sterically more crowded amine function of diamine I (Scheme 4) does not react with ketones to form ketimines, but only with aldehydes to form aldimines. On the other hand, the less crowded amine function does react with aldehydes as well as with ketones to form imines. Thus, the reaction of the diamine I with the triketone H affords chemoselectively the triplesalen ketimine half-unit J. In the second step, this triple ketimine J reacts with a simple salicylaldehyde to yield the desired triplesalen (Scheme 4).132 This stepwise protocol allows the use of variously substituted salicylaldehydes in the second step and the ligands H6talen, H6talent-Bu2, and H6talenNO2 have been reported.133 Besides these triplesalen ligands, a chiral version using the chiral diaminetrans-1,2-cyclohexanediamine,134,135 a triplesalophen ligand,136 and a triplesalacen ligand have been realized.137
 | ||
| Scheme 4 | ||
4. Properties of trinuclear triplesalen complexes
The trinucleating tris(tetradentate) triplesalen ligands H6talenX2 are capable to coordinate three transition metal ions. In a series of trinuclear nickel triplesalen complexes we have shown that variation of the terminal substituents in the triplesalen ligands provides a control of the electronic communication between the metal–salen subunits.133 The corresponding trinuclear copper triplesalen complexes all exhibit ferromagnetic interactions with St = 3/2 spin ground states.138,139EPR spectra display a ten line hyperfine splitting pattern due to the coupling of the total electron spin with three Cu nuclear spins.138 DFT calculations demonstrate that the spin-density on adjacent carbon atoms of the central benzene ring alters in the quartet ground state in accordance with the spin-polarization mechanism.138,139 The VIV ions in the trinuclear vanadyl complex [(talent-Bu2)(VIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)3] also reveal weak but ferromagnetic interactions.140
O)3] also reveal weak but ferromagnetic interactions.140
A common feature of the complexes with the tert-butyl derivative (talent-Bu2)6− is a severe ligand folding leading to an overall bowl-shaped structure. Interestingly, there is a driving force for two of these bowl-shaped trinuclear complexes to dimerize to disk-shaped hexanuclear assemblies. The interior of this dimer can be filled with non-covalently bound solvent molecules as observed in the complex [(talent-Bu2)Cu3] (Fig. 3a–c).139 The interior can also be filled with covalently bound molecules which bridge the metal ions of the individual trinuclear triplesalen complexes as in [{(talent-Bu2)Mn3(MeOH)}2(μ2-OAc)3]3+ (Fig. 3d). In this complex, two trinuclear MnIII triplesalen complexes are bridged by three acetates resulting in an overall hexanuclear MnIII6 complex.141 The driving force for dimerization seems to be so strong that entities are stabilized inside, which are not commonly detected as free species. The FeIII ions in the trinuclear FeIII triplesalen complex [(talent-Bu2){FeIII(MeOH)(H2O)}3]3+ are hexa-coordinate with one methanol molecule and one water molecule in the apical positions. The methanol molecules are placed inside the bowl (Fig. 3e). Two [(talent-Bu2){FeIII(MeOH)(H2O)}3]3+ units dimerize and encapsulate the uncommon [Cl⋯H⋯Cl]− species by interaction of the chloride ions with the coordinated methanol molecules (Fig. 3e).142 Another example is the stabilization of an unprecedented [V3O9]3− unit between two [(talent-Bu2){Mn(MeOH)}3]3+ building blocks (Fig. 3f).143
![Molecular structures of (a), (b) and (c) [(talent-Bu2)Cu3],139 (d) [{(talent-Bu2)Mn3(MeOH)}2(μ2-OAc)3]3+,141 (e) [{(talent-Bu2){FeIII(MeOH)(H2O)}3}2(ClHCl)]3+,142 (f) [{(talent-Bu2){Mn(MeOH)}3}2(V3O9)]3+.143](/image/article/2011/CC/c0cc02259d/c0cc02259d-f3.gif) | ||
| Fig. 3 Molecular structures of (a), (b) and (c) [(talent-Bu2)Cu3],139 (d) [{(talent-Bu2)Mn3(MeOH)}2(μ2-OAc)3]3+,141 (e) [{(talent-Bu2){FeIII(MeOH)(H2O)}3}2(ClHCl)]3+,142 (f) [{(talent-Bu2){Mn(MeOH)}3}2(V3O9)]3+.143 | ||
5. Heptanuclear complexes [MnIIIIII66M]3+3+
5.1 Synthesis and molecular structures
We have been inspired by the well-established chemistry of combining salen complexes with hexacyanometallates as bridging units, which results either in polynuclear complexes or in extended structures of 1D chains, 2D sheets, and 3D networks with interesting magnetic properties.17,29,38,39,144–148 However, despite the success of the combination of metal salen units with hexacyanometallates and the highly predictable exchange coupling through a CN− bridge,17,19,147 the nuclearity and dimensionality of these complexes are difficult to control.In this respect, we thought that the ligand folding in our trinuclear building blocks [(talent-Bu2){Mt(solv)n}3]m+ preorganizes the three Mt ions for coordination of three facial nitrogen atoms of a hexacyanometallate [Mt(CN)6]n−. Thus, a hexacyanometallate should bridge two trinuclear triplesalen building blocks in a hexa-connecting fashion (Fig. 4). We anticipated that the heptanuclear complexes [MnIIIIII66MIIIIII]3+3+ should fulfill all the requirements for SMMs, namely: (i) the triplesalen bridge favors a parallel spin alignment of the three MnIII ions of a molecular [(talent-Bu2)MnIII3]3+ building block, (ii) irrespective of the nature of the MnIII–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–M pathways (ferromagnetic or antiferromagnetic) a high spin ground state of the heptanuclear complex would occur as an efficient ferrimagnetic scheme arises from antiferromagnetic interactions, (iii) the [(salen)MnIII]+ units provide a pronounced single-site magnetic anisotropy, (iv) the C3 symmetry precludes that the single-site Dis compensate completely by projecting onto St, and (v) the molecular C3 axis enforces ESt = 0 thus minimizing the disturbing tunneling probability.
C–M pathways (ferromagnetic or antiferromagnetic) a high spin ground state of the heptanuclear complex would occur as an efficient ferrimagnetic scheme arises from antiferromagnetic interactions, (iii) the [(salen)MnIII]+ units provide a pronounced single-site magnetic anisotropy, (iv) the C3 symmetry precludes that the single-site Dis compensate completely by projecting onto St, and (v) the molecular C3 axis enforces ESt = 0 thus minimizing the disturbing tunneling probability.
![Building block approach for the construction of the heptanuclear complexes [Mccc66Mttt]3+3+ (Mt: terminal metal ion of the triplesalen building block; Mc: central metal ion of the hexacyanometallate). The symmetry of the trinuclear triplesalen building blocks is imposed on the molecular structure of the supramolecular assembly.](/image/article/2011/CC/c0cc02259d/c0cc02259d-f4.gif) | ||
| Fig. 4 Building block approach for the construction of the heptanuclear complexes [Mccc66Mttt]3+3+ (Mt: terminal metal ion of the triplesalen building block; Mc: central metal ion of the hexacyanometallate). The symmetry of the trinuclear triplesalen building blocks is imposed on the molecular structure of the supramolecular assembly. | ||
Our first reaction was that of [(talent-Bu2){MnIII(solv)n}3]3+ with [Fe(CN)6]3−. This was inspired by the work of Long et al., who demonstrated that already a trinuclear complex comprised of two MnIII salen complexes bridged by a central [FeIII(CN)6]3− acts as a SMM due to the ferromagnetic St = 9/2 spin ground state, the local magnetic anisotropy of the MnIII salen units, and the linear topology.149 Miyasaka and Clérac et al. used these trinuclear building blocks to form 1D chains, which exhibit single-chain magnetic (SCM) behavior.150 Indeed, we obtained the heptanuclear complex [{(talent-Bu2)MnIII3}2{FeIII(CN)6}]3+, [MnIIIIII66FeIIIIII]3+3+, isolated as its [Fe(CN)6]3− salt.151
The molecular structure of [MnIIIIII66FeIIIIII]3+3+ is illustrated in Fig. 5. Two trinuclear MnIII triplesalen units are bridged by a hexacyanoferrate. The approximate point group of this trication is S6. Although this was our first (synthesized and completely characterized in 2003) heptanuclear complex [Mttt66Mccc]n+nn+, it was published only in 2009,151 as the magnetic properties (vide infra) could only be understood and properly analyzed in a fruitful collaboration with Jürgen Schnack at Bielefeld University.
![Molecular structure of [MnIIIIII66FeIIIIII]3+3+ in various accentuations.151](/image/article/2011/CC/c0cc02259d/c0cc02259d-f5.gif) | ||
| Fig. 5 Molecular structure of [MnIIIIII66FeIIIIII]3+3+ in various accentuations.151 | ||
In analogy to the synthesis of [MnIIIIII66FeIIIIII]3+3+, we reacted [(talent-Bu2){MnIII(solv)n}3]3+ with [Cr(CN)6]3− and isolated the heptanuclear complex [{(talent-Bu2)MnIII3}2{CrIII(CN)6}]3+, [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+, as its (BPh4)− salt.152 In order to simplify the spin coupling scheme and reduce the number of parameters to simulate the magnetic properties, we synthesized the analogous heptanuclear complex [MnIIIIII66CoIIIIII]3+3+ including a diamagnetic CoIII l.s. ion.153 The molecular structures of the heptanuclear complexes [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ and [MnIIIIII66CoIIIIII]3+3+ are closely related to that of [MnIIIIII66FeIIIIII]3+3+.
5.2 Magnetic properties of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+
The magnetic properties of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ were the first we analyzed in detail and are therefore discussed here in the beginning. The temperature-dependence of the effective magnetic moment, μeff, of [MnIIIIII66CrIIIIII](BPh4)3 is typical for a ferrimagnetic coupling scheme with a minimum at 90 K (Fig. 6a) and a maximum of μeff = 17.75 μB at 5 K. This ferrimagnetic behavior is in accordance with an antiparallel orientation of the central CrIII spin (Si = 3/2) with the terminal MnIII spins (Si = 2) resulting in a St = 21/2 ground state. The variable-temperature and variable-field (VTVH) magnetization measurements with a saturation value at 7 T of 18.4 μB in combination with the nesting behavior of the isofield lines (Fig. 6b) demonstrate that [MnIIIIII66CrIIIIII](BPh4)3 possesses an anisotropic high spin ground state.152![(a) Temperature-dependence of μeff at 0.05 T and (b) VTVH at 1, 4, and 7 T for [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+.152 The solid line corresponds to a simulation based on parameters provided in the text.](/image/article/2011/CC/c0cc02259d/c0cc02259d-f6.gif) | ||
| Fig. 6 (a) Temperature-dependence of μeff at 0.05 T and (b) VTVH at 1, 4, and 7 T for [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+.152 The solid line corresponds to a simulation based on parameters provided in the text. | ||
In order to get an estimate of the exchange coupling constants, the temperature-dependence of μeff was simulated with a HDvV spin-Hamiltonian by full matrix-diagonalization. The topology of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ requires a coupling scheme with two different coupling constants: JMn–Cr describes the interaction between the central CrIII (S7) with each MnIII (S1 to S6) and JMn–Mn describes the interaction between two MnIII belonging to the same trinuclear triplesalen subunit. This results in the Hamiltonian specified in eqn (1).
| HHDvV = −2JMn–Cr(S1S7 + S2S7 + S3S7 + S4S7 + S5S7 + S6S7) − 2JMn–Mn(S1S2 + S2S3 + S3S1 + S4S5 + S5S6 + S6S4) | (1) |
Altogether, [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ fulfills all requirements for a SMM. It has a high spin ground state of St = 21/2 and the VTVH data indicate relatively strong magnetic anisotropy. Additionally, the molecular symmetry of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ is approximately C3. Indeed, ac susceptibility data exhibit a frequency-dependent out-of-phase signal, which is a typical signature for slow relaxation of the magnetization in accordance with [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ being a SMM. Finally, a field-dependent magnetization study on a single-crystal of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ exhibits hysteresis. Thus, [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ is a SMM and the anisotropy barrier Ueff was determined from the ac measurements to be 25.4 K.152
The reaction of [Cr(CN)6]3− with [(salen)Mn(OH2)]+ results in the formation of a closely related heptanuclear complex [{(salen)MnIII}6{Cr(CN)6}]3+ which exhibits also a high spin ferrimagnetic ground state of St = 21/2 due to antiferromagnetic interactions.155 Interestingly, while [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ is a SMM, [{(salen)MnIII}6{Cr(CN)6}]3+ is not a SMM.156 This difference can be related to the overall symmetry of the metal ion arrangements in these two complexes. While the six MnIII ions in [{(salen)MnIII}6{Cr(CN)6}]3+ are surrounding the central CrIII ion almost octahedrally (Fig. 7a, Mn–Mn distances 7.4 and 7.7 Å), the triplesalen causes a trigonal elongation (Fig. 7b, Mn–Mn distances 6.8 and 8.3 Å). Thus, the local anisotropy tensors in [{(salen)MnIII}6{Cr(CN)6}]3+ cancel each other upon projection onto the spin ground state while the trigonal elongation in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ prevents this cancellation. Additionally, the longer Mn–NNC bond distances of 2.32 Å in [{(salen)MnIII}6{Cr(CN)6}]3+ in comparison to 2.18 Å in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ should result in weaker exchange couplings and thus in a less well separated spin ground state.
![Comparison of the central Mn6Cr unit in the molecular structures of (a) [{(salen)MnIII}6{Cr(CN)6}]3+ 148 and (b) [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+.152](/image/article/2011/CC/c0cc02259d/c0cc02259d-f7.gif) | ||
Fig. 7 Comparison of the central Mn6Cr unit in the molecular structures of (a) [{(salen)MnIII}6{Cr(CN)6}]3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 148 and (b) [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+.152 148 and (b) [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+.152 | ||
5.3 Magnetic properties of [MnIIIIII66CoIIIIII]3+3+
The analysis of the magnetic properties of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ provided antiferromagnetic exchange couplings (JMn–Mn = −1.03 cm−1) between the MnIII ions of a trinuclear triplesalen building block. As this result was unexpected, a multitude of simulations using a ferromagnetic exchange coupling were performed but these did not reproduce the temperature-dependence of μeff. These antiferromagnetic interactions are in contradiction to the expected spin-polarization mechanism through the phloroglucinol backbone and to the experimentally observed ferromagnetic couplings in the trinuclear CuII and VIV complexes (see Section 4). In order to simplify the analysis of the magnetic data and to reduce the exchange pathways, we have synthesized the heptanuclear complex [MnIIIIII66CoIIIIII]3+3+ with a central diamagnetic CoIII low-spin ion. In collaboration with Jürgen Schnack, we have performed a full-matrix diagonalization of the appropriate spin-Hamiltonian including isotropic HDvV exchange, zero-field splitting, and Zeeman interaction.153 A frequently used simplification in such spin-Hamiltonians is a collinearity of the local D-tensors, which is usually not justified. We have incorporated the zero-field splitting for the MnIII ions including the relative orientations of the individual D-tensors by the angle ϑ of the Jahn–Teller axis with the molecular C3 axis. The continuous decrease of μeff with decreasing temperature (Fig. 8a) indicates dominant antiferromagnetic interactions and the strong nesting behavior of the VTVH magnetization data (Fig. 8b) indicates a strong magnetic anisotropy. Surprisingly, a satisfactory reproduction of the magnetic data required the incorporation not only of an exchange interaction between the MnIII ions belonging to one triplesalen subunit (JMn–Mn) but also of an exchange coupling between MnIII ions belonging to different triplesalen subunits (JMn–Mn′). A satisfactory reproduction of the experimental data has been obtained for the parameter set JMn–Mn = −(0.50 ± 0.04) cm−1, JMn–Mn′ = +(0.05 ± 0.02) cm−1, and D = −(2.5 ± 0.5) cm−1.153![(a) Temperature-dependence of μeff at 0.1 T and (b) VTVH at 1, 4, and 7 T for [MnIIIIII66CoIIIIII]3+3+.153 The solid lines correspond to simulations based on parameters provided in the text.](/image/article/2011/CC/c0cc02259d/c0cc02259d-f8.gif) | ||
| Fig. 8 (a) Temperature-dependence of μeff at 0.1 T and (b) VTVH at 1, 4, and 7 T for [MnIIIIII66CoIIIIII]3+3+.153 The solid lines correspond to simulations based on parameters provided in the text. | ||
This detailed analysis manifests that the exchange coupling through the phloroglucinol bridging unit is antiferromagnetic in nature for MnIII ions despite the ferromagnetic couplings observed in trinuclear CuII complexes92,138,139 and a VIVO complex.140 A crucial parameter for an efficient spin-polarization mechanism through the bridging benzene unit was found to be the amount of spin-density in the pzπ orbitals of the phenolic oxygen atoms.75 This spin-density depends on the metal ion, the remaining coordination sites, and on the ligand folding at the central metal–phenolate bond. Additionally, the trinuclear MnIII triplesalen building blocks have some degree of freedom to orient the three MnIII ions for optimal bonding to the facially arranged cyanide nitrogen atoms, which results in different orientations of the magnetic d orbitals relative to the phenolate p orbitals. Our studies on the trinuclear triplesalen complexes indicate that variations in these orientations can have a strong impact on the two different mechanisms, spin-polarization and spin-delocalization,75 and thus on the overall exchange interaction of the three MnIII ions through the phloroglucinol unit.
Additionally, a detailed analysis of the JMn–Mn coupling of the three available heptanuclear complexes [MnIIIIII66MIIIIII]3+3+ (MCr, Fe, Co) indicates that the exchange interaction between the MnIII ions belonging to the same triplesalen subunit involves not only an exchange pathway through the central phloroglucinol unit but also an exchange pathway through the central metal ion.153
5.4 Magnetic properties of [MnIIIIII66FeIIIIII]3+3+
The value of μeff of [MnIIIIII66FeIIIIII]3+3+ decreases monotonically with decreasing temperature (Fig. 9a) indicating dominant antiferromagnetic interactions between the metal ions.151 However, antiferromagnetic interactions between the central FeIII l.s. ion and the MnIII ions should result in an increase of μeff at lower temperatures due to a ferrimagnetic coupling scheme (see Section 5.2 for [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+). Therefore, the continuous decrease of μeff with decreasing temperature was already an indication that the contribution from magnetic anisotropy had a stronger impact on the overall magnetic properties than the exchange interactions. The magnetic data have been analyzed in detail with the methodology described for [MnIIIIII66CoIIIIII]3+3+. We have been aware that a perfect reproduction of the experimental data with one unique set of parameters is beyond the employed spin-Hamiltonian as other effects like non-negligible orbital effects on the exchange coupling in low-spin FeIII, the occurrence of intermolecular couplings, and the lower molecular symmetry in the solid state have not been considered. However, a reasonable range of parameters that reproduce both the temperature-dependent magnetization at 1 T and the VTVH magnetization data is given by: JMn–Mn = −(0.85 ± 0.15) cm−1, JFe–Mn = +(0.70 ± 0.30) cm−1, and DMn = −(3.0 ± 0.7) cm−1. The simulation of the 1 T data in Fig. 9b was much more difficult than that of the 4 T and 7 T data. This indicates the presence of intermolecular dipolar interactions between the large magnetic moments of [MnIIIIII66FeIIIIII]3+3+. The higher fields of 4 T and 7 T are able to break down these dipolar interactions more efficiently than the 1 T field. This is an important observation as stray fields from neighboring molecules can have the same effect as a rhombic term in the anisotropy with regard to the probability of the tunnelling through the anisotropy barrier. In this respect, even a pure C3 symmetric complex will always be influenced by its environment in the solid state, even at the large distances found in these heptanuclear complexes.![(a) Temperature-dependence of μeff at 1 T and (b) VTVH at 1, 4, and 7 T for [MnIIIIII66FeIIIIII]3+3+.151 The solid lines correspond to simulations based on parameters provided in the text.](/image/article/2011/CC/c0cc02259d/c0cc02259d-f9.gif) | ||
| Fig. 9 (a) Temperature-dependence of μeff at 1 T and (b) VTVH at 1, 4, and 7 T for [MnIIIIII66FeIIIIII]3+3+.151 The solid lines correspond to simulations based on parameters provided in the text. | ||
ac susceptibility data exhibit no out-of-phase signals above 1.9 K, demonstrating that [MnIIIIII66FeIIIIII]3+3+ is not a SMM. It is interesting to compare the magnetic data of [MnIIIIII66FeIIIIII]3+3+ to those of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ in order to obtain insight into why [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ is a SMM and [MnIIIIII66FeIIIIII]3+3+ is not a SMM. The coupling of the MnIII ions with the central CrIII ion in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ was found to be JCr–Mn = −5.0 cm−1. This results in the stabilization of a high-spin ground state of St = 21/2. On the other hand, the small coupling JFe–Mn = +0.70 cm−1 in [MnIIIIII66FeIIIIII]3+3+ results in a strong mixing of MS wave functions of various spin states, so that no high spin ground state is stabilized. Thus, the reason for [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ being a SMM and [MnIIIIII66FeIIIIII]3+3+ not being a SMM is the large JCr–Mn coupling compared to the small JFe–Mn coupling.
6. The tetranuclear complex [MnIIIIII33CrIIIIII]3+3+ as a half-unit: insight into the driving force for dimerization
In order to obtain more insight into the abstract driving force for dimerization of two trinuclear tripelsalen units, we synthesized as a half-unit of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ the tetranuclear complex [{(talent-Bu2)MnIII3}{(Me3tacn)Cr(CN)3}]3+, [MnIIIIII33CrIIIIII]3+3+ (Fig. 10), by reaction of the trinuclear MnIII triplesalen complex [(talent-Bu2){MnIII(solv)n}3]3+ with [(Me3tacn)Cr(CN)3].157 Several experimental observations indicate that [MnIIIIII33CrIIIIII]3+3+ has an overall lower stability than the heptanuclear [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+. Interestingly, this lower stability of [MnIIIIII33CrIIIIII]3+3+ is correlated with a significantly longer Mn–NNC bond length of 2.29 Å and a reduced ligand folding at the central phenolate of 35.8° in [MnIIIIII33CrIIIIII]3+3+ in comparison to 2.18 Å and 46.7° in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+, respectively.
![Molecular structure of [MnIIIIII33CrIIIIII]3+3+.157](/image/article/2011/CC/c0cc02259d/c0cc02259d-f10.gif) | ||
| Fig. 10 Molecular structure of [MnIIIIII33CrIIIIII]3+3+.157 | ||
VTVH and μeffvs. T magnetic data have been analyzed in detail by full-matrix diagonalization of the appropriate spin-Hamiltonian, consisting of isotropic exchange, zero-field splitting, and Zeeman interaction components. Satisfactory reproduction of the experimental data has been obtained for the parameters JMn–Cr = −(0.12 ± 0.04) cm−1, JMn–Mn = −(0.70 ± 0.03) cm−1, and DMn = −(3.0 ± 0.4) cm−1. These generate a triply-degenerate pseudo St = 7/2 spin manifold, which cannot be appropriately described by a giant spin model and which exhibits a weak easy-axis magnetic anisotropy. This is corroborated by the onset of a frequency-dependent χ″ signal at low temperatures, demonstrating a slow relaxation of the magnetization indicative of [MnIIIIII33CrIIIIII]3+3+ being a SMM.
The higher driving force for the [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ formation is not only correlated with a shorter Mn–NNC bond length but also with a stronger antiferromagnetic JMn–Cr exchange coupling of −5.0 cm−1vs. −0.12 cm−1 in [MnIIIIII33CrIIIIII]3+3+. A detailed analysis in conjunction with magnetic properties of complexes with a MnIII–NC–CrIII exchange pathway found in the literature and a close inspection of the molecular structures of the heptanuclear complexes [Mttt66Mccc]n+nn+ (see space-filling model of [MnIIIIII66FeIIIIII]3+3+ in Fig. 11) identifies some kind of van der Waals-type contacts between the two trinuclear triplesalen building blocks. In this respect, the absence of a second triplesalen subunit in [MnIIIIII33CrIIIIII]3+3+ may reduce the driving force for ligand folding. The stronger supramolecular attraction between the two [(talent-Bu2)MnIII3]3+ subunits in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ enforces a more substantial bending, which results in shorter Mn–NNC bond lengths and therefore higher Mn–NNC bond strengths. This leads to the observed high driving force for [MnIIIIII66MIIIIII]3+3+ formation and thus to the strong JMn–Cr coupling, which is the origin of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ being a good SMM.
![Molecular structure of [MnIIIIII66FeIIIIII]3+3+ in space-filling representation with the two trinuclear triplesalen building blocks colored individually to illustrate van der Waals interactions between the molecular building blocks.](/image/article/2011/CC/c0cc02259d/c0cc02259d-f11.gif) | ||
| Fig. 11 Molecular structure of [MnIIIIII66FeIIIIII]3+3+ in space-filling representation with the two trinuclear triplesalen building blocks colored individually to illustrate van der Waals interactions between the molecular building blocks. | ||
7. Conclusions and outlook
Based on the extensive work on Mn121212OAc and other SMMs, we have formulated the requirements for the rational design of new families of SMMs. Besides a high-spin ground state St and a strong magnetic anisotropy of the easy-axis type, careful considerations of the overall symmetry of the complexes should be made. The symmetry should be at least C3 to minimize rhombic components in the anisotropy and therefore the tunnelling probability through the anisotropy barrier, but lower than cubic to avoid a cancellation of the local anisotropies upon projection onto the spin ground state.Our approach is based on concepts of supramolecular chemistry including topological constraints, complementarity, preorganization, and molecular recognition, where the prerequisites for SMMs are programmed into a directional polynucleating ligand of low flexibility. In this respect, we have designed the hybrid ligand triplesalen, which combines a phloroglucinol bridging unit to obtain ferromagnetic interactions by the spin-polarization mechanism with a salen-like coordination environment to introduce a strong tetragonal ligand-field component for magnetic anisotropy.
We have shown in a series of trinuclear triplesalen complexes132–134,138,139,158 that (i) the C3 symmetry of the triplesalen ligand is imposed on the complex, (ii) phloroglucinol transmits weak but ferromagnetic interactions between CuII and VIV, and (iii) a severe ligand folding occurs in [(talent-Bu2)M3], resulting in an overall bowl-shaped molecular structure.133,139
We have taken advantage of the ligand folding in the triplesalen complexes by reacting two trinuclear MnIII triplesalen complexes [(talent-Bu2){MnIII(solv)n}3]3+ as molecular building blocks with [M(CN)6]3− as a hexaconnector (Fig. 4) and obtained the isostructural series [MnIIIIII66FeIIIIII]3+3+, [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+, and [MnIIIIII66CoIIIIII]3+3+ with [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ being a SMM.
The analysis of the magnetic properties provides an antiferromagnetic coupling between the MnIII ions through the phloroglucinol ring, which is especially evident in [MnIIIIII66CoIIIIII]3+3+ with a central diamagnetic CoIII l.s. ion. The strong antiferromagnetic interaction in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ between the central CrIII and the terminal MnIII ions (JCr–Mn = −5.0 cm−1) stabilizes an St = 21/2 spin ground state, while the coupling in [MnIIIIII66FeIIIIII]3+3+ (JFe–Mn = +0.7 cm−1) is too weak to isolate a single spin ground state. Therefore, [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ is a SMM, while [MnIIIIII66FeIIIIII]3+3+ is not. Comparison of the properties of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ and the tetranuclear [MnIIIIII33CrIIIIII]3+3+ half-unit established a shortening of the Mn–NCN bond from 2.29 Å in [MnIIIIII33CrIIIIII]3+3+ to 2.18 Å in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ by the attractive van der Waals interactions between the two triplesalen subunits. These attractive forces are thus responsible for the stronger coupling of −5.0 cm−1 in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ compared to −0.12 cm−1 in [MnIIIIII33CrIIIIII]3+3+.
Although [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ is a rationally developed SMM, the question remains as to why the anisotropy barrier is not higher. Our current research identifies several crucial points, which must be improved in order to obtain a higher anisotropy barrier. In this respect, it must be regarded as the main advantage that the modular assembly of the heptanuclear complexes from three molecular building blocks allows the fine-tuning of the molecular and steric properties of each building block without losing the driving force for the formation of the heptanuclear complexes.
We have synthesized, structurally and magnetically characterized an isostructural series of [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ SMMs differing in the counterion and/or the solvent molecules of crystallization.154 An important parameter, which has strong impact on the magnetic properties, turned out to be the ligand at the sixth coordination site of the MnIII ions. MnIII salen units are usually 5- or 6-coordinate with a very flat potential for the coordination of a solvent molecule at the sixth position. However, the presence or absence of a solvent molecule such as methanol or acetonitrile at this position influences the Mn–NNC distance and therefore the coupling constant. We have obtained [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ complexes, where the sixth positions are all occupied by methanol molecules, which results in a good approximation of molecular C3 symmetry, while others have a mixture of 5-coordinate and 6-coordinate MnIII ions.
Another structural aspect is the orientation of the neighboring [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ molecules in the crystal structure. The detailed investigation of the magnetic properties indicates that there are intermolecular, dipolar interactions despite the large intermolecular distances. These dipolar fields of neighboring molecules act as stray fields and introduce some rhombic terms in the magnetic anisotropy. As stated already in Section 2.6, rational design does not mean that structural parameters can be varied to a preferred value. In this respect, the nature of the sixth coordination sites of the MnIII ion and the packing of the molecules in the crystal are difficult to optimize.
Another handle to increase the anisotropy barrier in [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+ is a better stabilization of the spin ground state. An overlap of the spin multiplets leads to a mixing of MS states belonging to different spin-multiplets. This must be regarded as a source for the effective barrier Ueff being usually smaller than the calculated anisotropy barrier U = DSt·St2. Therefore, we are trying to optimize the triplesalen ligand backbone in order to increase the contributions of the spin-polarization mechanism over those of the spin-delocalization mechanism. In this respect, we are trying to change the central 1,3,5-trihydroxybenzene bridging unit to a 1,3,5-trimercaptobenzene bridging unit, as metal–sulfur bonds are more covalent leading to stronger spin densities in the sulfur pzπ orbitals.159
Another approach to improve the spin-polarization mechanism is based on the observation that the central 1,3,5-trihydroxybenzene unit in our ligands and our complexes has lost a large degree of their aromaticity and a resonance structure corresponding to a radialene-like resonance hybrid has strong contributions.160 This provides us with a synthetic handle to increase the aromaticity in the central unit by replacing the imine units with amine units.
The projection of magnetic anisotropy tensors onto the spin ground state is maximal for a collinear arrangement of the anisotropy tensors. In this respect, we have synthesized the corresponding triplesalophene ligand, which has a completely sp2-hybridized ligand backbone,136 which should enforce a planar trinuclear building block and therefore collinear MnIII anisotropy tensors. Such planar molecular building blocks should enable the construction of triple-stranded nonanuclear complexes [Mttt33Mccc33Mttt33]n+nn+ or triple-stranded 1D chains.
Besides changing the triplesalen ligand, the driving force for the formation of the heptanuclear assemblies also allows the variation of the terminal metal ions and of the central hexacyanometallate. In this respect, we have been able to synthesize the isostructural complexes [MnIIIIII66MnIIIIII]3+3+, [FeIIIIII66CrIIIIII]3+3+, [FeIIIIII66MnIIIIII]3+3+, [FeIIIIII66FeIIII]2+2+, [MnIIIIII66FeIIII]2+2+ as well as the 4d homologue [MnIIIIII66MoIIIIII]3+3+ and the 5d homologue [MnIIIIII66OsIIIIII]3+3+. Additionally, we are changing the nature of the terminal donor atoms in triplesalen to stabilize also low-valent metal ions like CoII or VIII, which might be interesting with respect to the magnetic properties of the heptanuclear complexes. We hope that a single modification or a combination of such modifications will allow us to rationally improve the magnetic properties compared to the existing SMM [MnIIIIIIIIIIII6666CrIIIIIIIIIIII]3+3+3+3+.
Acknowledgements
I am grateful to all my coworkers on this project whose names can be found in the references. Especially their enthusiasm for this project, despite severe problems in the synthesis of molecules developed on a piece of paper, has been central for the success of this work. This work was supported by the DFG (SFB 424, FOR945 ‘Nanomagnets: from Synthesis via Interactions with Surfaces to Function’), the Fonds der Chemischen Industrie, and Bielefeld University.Notes and references
- O. Kahn, Molecular Magnetism, VCH Publisher, New York, 1993 Search PubMed.
- See special issue ‘Forum on Molecular Magnetism’ of Inorg. Chem. 2009, 48(8).
- See ‘Issue on Molecular Magnets’ of Dalton Trans. 2010, 39(20).
- D. Gatteschi, O. Kahn, J. S. Miller and F. Palacio, Adv. Mater., 1991, 3, 161–163 CrossRef.
- J. S. Miller and A. J. Epstein, Angew. Chem., Int. Ed. Engl., 1994, 33, 385–415 CrossRef.
- Molecular Magnetism: From Molecular Assemblies to the Devices, ed. E. Coronado, P. Delhaès, D. Gatteschi and J. S. Miller, Kluwer Academic Publishers, Dordrecht, 1996 Search PubMed.
- J. S. Miller and M. Drillon, Magnetism: Molecules to Materials, Wiley-VCH, Weinheim, 2001–2005, vol. I–IV Search PubMed.
- J. S. Miller, Inorg. Chem., 2000, 39, 4392–4408 CrossRef CAS.
- G. A. Timco, S. Carretta, F. Troiani, F. Tuna, R. J. Pritchard, C. A. Muryn, E. J. L. McInnes, A. Ghirri, A. Candini, P. Santini, G. Amoretti, M. Affronte and R. E. P. Winpenny, Nat. Nanotechnol., 2009, 4, 173–178 CrossRef CAS.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS.
- J. S. Miller, J. C. Calabrese, A. J. Epstein, R. W. Bigelow, J. H. Zhang and W. M. Reiff, J. Chem. Soc., Chem. Commun., 1986, 1026–1028 RSC.
- J. S. Miller, J. C. Calabrese, H. Rommelmann, S. R. Chittipeddi, J. H. Zhang, W. M. Reiff and A. J. Epstein, J. Am. Chem. Soc., 1987, 109, 769–781 CrossRef CAS.
- J. M. Manriquez, G. T. Yee, R. S. McLean, A. J. Epstein and J. S. Miller, Science, 1991, 252, 1415–1417 CrossRef CAS.
- S. M. Holmes and G. S. Girolami, J. Am. Chem. Soc., 1999, 121, 5593–5594 CrossRef CAS.
- S. Ferlay, T. Mallah, R. Ouahes, P. Veillet and M. Verdaguer, Nature, 1995, 378, 701–703 CrossRef CAS.
- E. Dujardin, S. Ferlay, X. Phan, C. Desplanches, C. Cartier dit Moulin, P. Sainctavit, F. Baudelet, E. Dartyge, P. Veillet and M. Verdaguer, J. Am. Chem. Soc., 1998, 120, 11347–11352 CrossRef CAS.
- M. Verdaguer, A. Bleuzen, V. Marvaud, J. Vaissermann, M. Seuleiman, C. Desplanches, A. Scuiller, C. Train, R. Garde, G. Gelly, C. Lomenech, I. Rosenman, P. Veillet, C. Cartier and F. Villain, Coord. Chem. Rev., 1999, 190–192, 1023–1047 CrossRef CAS.
- T. Mallah, S. Thiebaut, M. Verdaguer and P. Veillet, Science, 1993, 262, 1554–1557 CrossRef CAS.
- H. Weihe and H. U. Güdel, Comments Inorg. Chem., 2000, 22, 75 CrossRef CAS.
- W. R. Entley, C. R. Treadway and G. S. Girolami, Mol. Cryst. Liq. Cryst., 1995, 273, 153–166 CrossRef.
- J. B. Goodenough, Magnetism and the Chemical Bond, Interscience, New York, 1963 Search PubMed.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98 CrossRef CAS.
- P. W. Anderson, Phys. Rev., 1959, 115, 2–13 CrossRef CAS.
- T. Lis, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2042–2046 CrossRef.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- R. Sessoli, H. L. Tsai, A. R. Schake, S. Y. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS.
- S. M. J. Aubin, M. W. Wemple, D. M. Adams, H. L. Tsai, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1996, 118, 7746–7754 CrossRef CAS.
- G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66–71 CAS.
- J. R. Long, in Chemistry of Nanostructured Materials, ed. P. Yang, World Scientific, Hong Kong, 2003, pp. 291–315 Search PubMed.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 Search PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS.
- J. Tejada, Polyhedron, 2001, 20, 1751–1756 CrossRef CAS.
- G. Aromi and E. K. Brechin, Struct. Bonding, 2006, 122, 1–67 CAS.
- M. Murrie, Chem. Soc. Rev., 2010, 39, 1986–1995 RSC.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Clarendon Press, Oxford, GB, 1970 Search PubMed.
- R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 1–10 RSC.
- L. M. C. Beltran and J. R. Long, Acc. Chem. Res., 2005, 38, 325–334 CrossRef CAS.
- J.-N. Rebilly and T. Mallah, Struct. Bonding, 2006, 122, 103–131 CAS.
- O. Roubeau and R. Clérac, Eur. J. Inorg. Chem., 2008, 4325–4342 CrossRef CAS.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- H.-L. Tsai, D.-M. Chen, C.-I. Yang, T.-Y. Jwo, C.-S. Wur, G.-H. Lee and Y. Wang, Inorg. Chem. Commun., 2001, 4, 511–514 CrossRef CAS.
- A. Cornia, R. Sessoli, L. Sorace, D. Gatteschi, A. L. Barra and C. Daiguebonne, Phys. Rev. Lett., 2002, 89, 257201 CrossRef CAS.
- W. Wernsdorfer, M. Murugesu and G. Christou, Phys. Rev. Lett., 2006, 96, 057208 CrossRef CAS.
- N. E. Chakov, S.-C. Lee, A. G. Harter, P. L. Kuhns, A. P. Reyes, S. O. Hill, N. S. Dalal, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2006, 128, 6975–6989 CrossRef CAS.
- E. Burzuri, C. Carbonera, F. Luis, D. Ruiz-Molina, C. Lampropoulos and G. Christou, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224428 CrossRef.
- S. M. J. Aubin, N. R. Dilley, M. W. Wemple, N. B. Maple, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1998, 120, 839–840 CrossRef CAS.
- N. Aliaga-Alcalde, R. S. Edwards, S. O. Hill, W. Wernsdorfer, K. Folting and G. Christou, J. Am. Chem. Soc., 2004, 126, 12503–12516 CrossRef CAS.
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS.
- A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clérac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., 2006, 118, 5048–5051 CrossRef.
- O. Waldmann, A. M. Ako, H. U. Güdel and A. K. Powell, Inorg. Chem., 2008, 47, 3486–3488 CrossRef CAS.
- A. M. Ako, V. Mereacre, R. Clérac, W. Wernsdorfer, I. J. Hewitt, C. E. Anson and A. K. Powell, Chem. Commun., 2009, 544–546 RSC.
- T. Glaser, I. Liratzis, A. M. Ako and A. Powell, Coord. Chem. Rev., 2009, 253, 2296–2305 CrossRef CAS.
- C. J. Milios, C. P. Raptopoulou, A. Terzis, F. Lloret, R. Vicente, S. P. Perlepes and A. Escuer, Angew. Chem., Int. Ed., 2004, 43, 210–212 CrossRef CAS.
- C. J. Milios, A. Vinslava, A. G. Whittaker, S. Parsons, W. Wernsdorfer, G. Christou, S. P. Perlepes and E. K. Brechin, Inorg. Chem., 2006, 45, 5272–5274 CrossRef CAS.
- C. J. Milios, A. Vinslava, W. Wernsdorfer, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 2754–2755 CrossRef CAS.
- C. J. Milios, R. Inglis, R. Bagai, W. Wernsdorfer, A. Collins, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, Chem. Commun., 2007, 3476–3478 RSC.
- C. J. Milios, R. Inglis, A. Vinslava, R. Bagai, W. Wernsdorfer, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 12505–12511 CrossRef CAS.
- S. Piligkos, J. Bendix, C. J. Milios and E. K. Brechin, Dalton Trans., 2008, 2277–2284 RSC.
- O. Kahn, Acc. Chem. Res., 2000, 33, 647–657 CrossRef CAS.
- C. Zener, Phys. Rev., 1951, 82, 403–405 CrossRef CAS.
- G. Blondin and J.-J. Girerd, Chem. Rev., 1990, 90, 1359–1376 CrossRef CAS.
- T. Glaser, K. Rose, S. E. Shadle, B. Hedman, K. E. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 442–454 CrossRef CAS.
- T. Beissel, F. Birkelbach, E. Bill, T. Glaser, F. Kesting, C. Krebs, T. Weyhermüller, K. Wieghardt, C. Butzlaff and A. X. Trautwein, J. Am. Chem. Soc., 1996, 118, 12376–12390 CrossRef CAS.
- T. Glaser and K. Wieghardt, ACS Symp. Ser., 1998, 692, 314–331 CAS.
- T. Glaser, T. Beissel, E. Bill, T. Weyhermüller, V. Schünemann, W. Meyer-Klaucke, A. X. Trautwein and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 2193–2208 CrossRef CAS.
- T. Glaser, F. Kesting, T. Beissel, E. Bill, T. Weyhermüller, W. Meyer-Klaucke and K. Wieghardt, Inorg. Chem., 1999, 38, 722–732 CrossRef CAS.
- O. Kahn, Inorg. Chim. Acta, 1982, 62, 3–14 CrossRef CAS.
- T. Glaser, H. Theil, I. Liratzis, T. Weyhermüller and E. Bill, Inorg. Chem., 2006, 45, 4889–4891 CrossRef CAS.
- H. C. Longuet-Higgins, J. Chem. Phys., 1950, 18, 265–274 CrossRef CAS.
- H. M. McConnell, J. Chem. Phys., 1963, 39, 1910 CrossRef CAS.
- J. Cano, E. Ruiz, S. Alvarez and M. Verdaguer, Comments Inorg. Chem., 1998, 20, 27–56 CrossRef CAS.
- J. A. McCleverty and M. D. Ward, Acc. Chem. Res., 1998, 31, 842–851 CrossRef CAS.
- D. A. Dougherty, Acc. Chem. Res., 1991, 24, 88–94 CrossRef CAS.
- T. Glaser, H. Theil and M. Heidemeier, C. R. Chim., 2008, 11, 1121–1136 CrossRef CAS.
- H. Iwamura, Adv. Phys. Org. Chem., 1991, 26, 179–253.
- A. Rajca, Chem.–Eur. J., 2002, 8, 4834–4841 CrossRef CAS.
- P. Karafiloglou, J. Chem. Phys., 1985, 82, 3728–3740 CrossRef CAS.
- M. Jung, A. Sharma, D. Hinderberger, S. Braun, U. Schatzschneider and E. Rentschler, Inorg. Chem., 2009, 48, 7244–7250 CrossRef CAS.
- S.-i. Mitsubori, T. Ishida, T. Nogami and H. Iwamura, Chem. Lett., 1994, 285–288 CAS.
- T. Ishida, S.-i. Mitsubori, T. Nogami, N. Takeda, M. Ishikawa and H. Iwamura, Inorg. Chem., 2001, 40, 7059–7064 CrossRef CAS.
- F. Lloret, G. De Munno, M. Julve, J. Cano, R. Ruiz and A. Caneschi, Angew. Chem., Int. Ed., 1998, 37, 135–138 CrossRef CAS.
- A. M. W. Cargill Thompson, D. Gatteschi, J. A. McCleverty, J. A. Navas, E. Rentschler and M. D. Ward, Inorg. Chem., 1996, 35, 2701–2703 CrossRef.
- L. C. Francesconi, D. R. Corbin, D. N. Hendrickson and G. D. Stucky, Inorg. Chem., 1979, 18, 3074–3080 CrossRef CAS.
- H. Oshio, J. Chem. Soc., Chem. Commun., 1991, 240–241 RSC.
- V. A. Ung, S. M. Couchman, J. C. Jeffery, J. A. McCleverty, M. D. Ward, F. Totti and D. Gatteschi, Inorg. Chem., 1999, 38, 365–369 CrossRef.
- I. Fernández, R. Ruiz, J. Faus, M. Julve, F. Lloret, J. Cano, X. Ottenwaelder, Y. Journaux and C. Munoz, Angew. Chem., Int. Ed., 2001, 40, 3039–3042 CrossRef CAS.
- E. Pardo, R. Ruiz-Garcia, J. Cano, X. Ottenwaelder, R. Lescouezec, Y. Journaux, F. Lloret and M. Julve, Dalton Trans., 2008, 2780–2805 RSC.
- X. Ottenwaelder, J. Cano, Y. Journaux, E. Riviere, C. Brennan, M. Nierlich and R. Ruiz-Garcia, Angew. Chem., Int. Ed., 2004, 43, 850–852 CrossRef CAS.
- M.-C. Dul, X. Ottenwaelder, E. Pardo, R. Lescouezec, Y. Journaux, L. M. Chamoreau, R. Ruiz-Garcia, J. Cano, M. Julve and F. Lloret, Inorg. Chem., 2009, 48, 5244–5249 CrossRef CAS.
- V. A. Ung, A. Thompson, D. A. Bardwell, D. Gatteschi, J. C. Jeffery, J. A. McCleverty, F. Totti and M. D. Ward, Inorg. Chem., 1997, 36, 3447–3454 CrossRef.
- T. Glaser, M. Gerenkamp and R. Fröhlich, Angew. Chem., Int. Ed., 2002, 41, 3823–3825 CrossRef CAS.
- A. Bencini and D. Gatteschi, Electron Paramagnetic Resonance of Exchanged Coupled Systems, Springer-Verlag, Berlin, 1990 Search PubMed.
- A. Bencini, I. Ciofini and M. G. Uytterhoeven, Inorg. Chim. Acta, 1998, 274, 90–101 CrossRef CAS.
- D. Gatteschi and L. Sorace, J. Solid State Chem., 2001, 159, 253–261 CrossRef CAS.
- B. N. Figgis, Introduction to Ligand Fields, Interscience Publishers, New York, 1964 Search PubMed.
- F. Neese and E. I. Solomon, in Magnetism: Molecules to Materials, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2003, vol. 4, pp. 345–466 Search PubMed.
- S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420–421 CrossRef CAS.
- C. M. Zaleski, E. C. Depperman, J. W. Kampf, M. L. Kirk and V. L. Pecoraro, Angew. Chem., Int. Ed., 2004, 43, 3912–3914 CrossRef CAS.
- R. Sessoli and A. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS.
- M. T. Gamer, Y. Lan, P. W. Roesky, A. Powell and R. Clérac, Inorg. Chem., 2008, 47, 6581–6583 CrossRef CAS.
- C. M. Zaleski, J. W. Kampf, T. Mallah, M. L. Kirk and V. L. Pecoraro, Inorg. Chem., 2007, 46, 1954–1956 CrossRef CAS.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS.
- S. Takamatsu, T. Ishikawa, S. Koshihara and N. Ishikawa, Inorg. Chem., 2007, 46, 7250–7252 CrossRef.
- V. Vitali, S. Fabris, A. M. Conte, S. Brink, M. Ruben, S. Baroni and K. Kern, Nano Lett., 2008, 8, 3364–3368 CrossRef CAS.
- S. Mitra, Prog. Inorg. Chem., 1977, 22, 309–408 CAS.
- R. Boca, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef CAS.
- B. J. Kennedy and K. S. Murray, Inorg. Chem., 1985, 24, 1552–1557 CrossRef CAS.
- L. B. Dugad, D. V. Behere, V. R. Marathe and S. Mitra, Chem. Phys. Lett., 1984, 104, 353–356 CrossRef CAS.
- W. Wernsdorfer and R. Sessoli, Science, 1999, 284, 133–135 CrossRef CAS.
- W. Wernsdorfer, Adv. Chem. Phys., 2001, 118, 99–190 CAS.
- L. Thomas, F. Lionti, R. Ballou, D. Gatteschi, R. Sessoli and B. Barbara, Nature, 1996, 383, 145–147 CrossRef CAS.
- J. R. Friedman, M. P. Sarachik, J. Tejada and R. Ziolo, Phys. Rev. Lett., 1996, 76, 3830–3833 CrossRef CAS.
- J.-M. Lehn, J. Inclusion Phenom., 1988, 6, 351–396 CrossRef CAS.
- D. J. Cram, J. Inclusion Phenom., 1988, 6, 397–413 CrossRef CAS.
- D. H. Busch, Chem. Rev., 1993, 93, 847–860 CrossRef CAS.
- K. A. Campbell, M. R. Lashley, J. K. Wyatt, M. H. Nantz and R. D. Britt, J. Am. Chem. Soc., 2001, 123, 5710–5719 CrossRef CAS.
- J. Krzystek and J. Telser, J. Magn. Reson., 2003, 162, 454–465 CrossRef CAS.
- H. Miyasaka, R. Clerac, W. Wernsdorfer, L. Lecren, C. Bonhomme, K.-i. Sugiura and M. Yamashita, Angew. Chem., Int. Ed., 2004, 43, 2801–2805 CrossRef CAS.
- C. Coulon, H. Miyasaka and R. Clérac, Struct. Bonding, 2006, 122, 163–206 CAS.
- R. Clerac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837–12844 CrossRef CAS.
- A. W. Kleij, Eur. J. Inorg. Chem., 2009, 193–205 CrossRef CAS.
- R. Hernández-Molina and A. Mederos, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Ltd., Oxford, vol. 1, 2004, pp. 411–446 Search PubMed.
- J. Lopez, S. Liang and X. R. Bu, Tetrahedron Lett., 1998, 39, 4199–4202 CrossRef CAS.
- K. B. M. Janssen, I. Laquierra, W. Dehaen, R. F. Parton, I. F. J. Vankelecom and P. A. Jacobs, Tetrahedron: Asymmetry, 1997, 8, 3481–3487 CrossRef CAS.
- J.-P. Costes, F. Dahan, A. Dupuis and J.-P. Laurant, J. Chem. Soc., Dalton Trans., 1998, 1307–1314 RSC.
- A. Böttcher, H. Elias, B. Eisenmann, E. Hilms, A. Huer, R. Kniep and C. Röhr, Z. Naturforsch., B: Chem. Sci., 1994, 49, 1089–1100.
- S. Dayagi and Y. Degani, in The Chemistry of the Carbon–Nitrogen Double Bond, ed. S. Patai, Interscience Publishers, London, 1970, p. 64 Search PubMed.
- R. Atkins, G. Brewer, E. Kokot, G. M. Mockler and E. Sinn, Inorg. Chem., 1985, 24, 127–134 CrossRef CAS.
- M. I. Fernandey Garcia, M. Fondo, A. M. Garcia Deibe, M. B. Fernandez Fernandez and A. M. Gonzalez, Z. Anorg. Allg. Chem., 2000, 626, 1985–1991 CrossRef.
- T. Glaser, M. Heidemeier, E. Krickemeyer and H. Bögge, Z. Naturforsch., B: Chem. Sci., 2006, 61, 753–757 CAS.
- T. Glaser, M. Heidemeier and T. Lügger, Dalton Trans., 2003, 2381–2383 RSC.
- T. Glaser, M. Heidemeier, R. Fröhlich, P. Hildebrandt, E. Bothe and E. Bill, Inorg. Chem., 2005, 44, 5467–5482 CrossRef CAS.
- C. Mukherjee, A. Stammler, H. Bögge and T. Glaser, Inorg. Chem., 2009, 48, 9476–9484 CrossRef CAS.
- C. Mukherjee, A. Stammler, H. Bögge and T. Glaser, Chem.–Eur. J., 2010, 16, 10137–10149 CrossRef CAS.
- G.-C. Frhr. v. Richthofen, A. Stammler, H. Bögge, T. Glaser, manuscript submitted for publication.
- S. Walleck, H. Theil, M. Heidemeier, G. Heinze-Brückner, A. Stammler, H. Bögge and T. Glaser, Inorg. Chim. Acta, 2010 DOI:10.1016/j.ica.2010.06.054.
- T. Glaser, M. Heidemeier, S. Grimme and E. Bill, Inorg. Chem., 2004, 43, 5192–5194 CrossRef CAS.
- T. Glaser, M. Heidemeier, J. B. H. Strautmann, H. Bögge, A. Stammler, E. Krickemeyer, R. Huenerbein, S. Grimme, E. Bothe and E. Bill, Chem.–Eur. J., 2007, 13, 9191–9206 CrossRef CAS.
- H. Theil, G.-C. Frhr. v. Richthofen, A. Stammler, H. Bögge and T. Glaser, Inorg. Chim. Acta, 2008, 361, 916–924 CrossRef CAS.
- T. Glaser, M. Heidemeier, H. Theil, A. Stammler, H. Bögge and J. Schnack, Dalton Trans., 2010, 39, 192–199 RSC.
- T. Glaser, M. Heidemeier, E. Krickemeyer, V. Hoeke, unpublished results.
- T. Glaser, E. Krickemeyer, V. Hoeke, unpublished results.
- H. Miyasaka, N. Matsumoto, H. Okawa, N. Re, E. Gallo and C. Floriani, J. Am. Chem. Soc., 1996, 118, 981–994 CrossRef CAS.
- N. Re, R. Crescenzi, C. Floriani, H. Miyasaka and N. Matsumoto, Inorg. Chem., 1998, 37, 2717–2722 CrossRef CAS.
- M. Ohba and H. Okawa, Coord. Chem. Rev., 2000, 198, 313–328 CrossRef CAS.
- K. R. Dunbar and R. A. Heintz, Prog. Inorg. Chem., 1996, 45, 283–391.
- A. V. Palii, S. M. Ostrovsky, S. I. Klokishner, B. S. Tsukerblat, C. P. Berlinguette, K. R. Dunbar and J. R. Galán-Mascarós, J. Am. Chem. Soc., 2004, 126, 16860–16867 CrossRef CAS.
- H. J. Choi, J. J. Sokol and J. R. Long, Inorg. Chem., 2004, 43, 1606–1608 CrossRef CAS.
- M. Ferbinteanu, H. Miyasaka, W. Wernsdorfer, K. Nakata, K.-i. Sugiura, M. Yamashita, C. Coulon and R. Clerac, J. Am. Chem. Soc., 2005, 127, 3090–3099 CrossRef CAS.
- T. Glaser, M. Heidemeier, E. Krickemeyer, H. Bögge, A. Stammler, R. Fröhlich, E. Bill and J. Schnack, Inorg. Chem., 2009, 48, 607–620 CrossRef CAS.
- T. Glaser, M. Heidemeier, T. Weyhermüller, R.-D. Hoffmann, H. Rupp and P. Müller, Angew. Chem., Int. Ed., 2006, 45, 6033–6037 CrossRef CAS.
- E. Krickemeyer, V. Hoeke, A. Stammler, H. Bögge, J. Schnack and T. Glaser, Z. Naturforsch., B: Chem. Sci., 2010, 65, 295–303 CAS.
- V. Hoeke, E. Krickemeyer, M. Heidemeier, J. Schnack, A. Stammler, H. Bögge, T. Glaser, manuscript in preparation.
- X. P. Shen, B. L. Li, J. Z. Zou, Z. Xu, Y. P. Yu and S. X. Liu, Transition Met. Chem., 2002, 27, 372 CrossRef CAS.
- H. J. Choi, J. J. Sokol and J. R. Long, J. Phys. Chem. Solids, 2004, 65, 839–844 CrossRef CAS.
- G.-C. Frhr. v. Richthofen, A. Stammler, H. Bögge, M. W. DeGroot, J. R. Long and T. Glaser, Inorg. Chem., 2009, 48, 10165–10176 CrossRef.
- T. Glaser, M. Heidemeier and R. Fröhlich, C. R. Chim., 2007, 10, 71–78 CrossRef CAS.
- H. Theil, B. Feldscher, A. Stammler, H. Bögge and T. Glaser, unpublished results.
- B. Feldscher, A. Stammler, H. Bögge and T. Glaser, Dalton Trans. 10.1039/c0dt00961j , in press.
Footnote |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| This journal is © The Royal Society of Chemistry 2011 |