New synthetic discoveries via high-pressure solid-state chemistry†
Hubert
Huppertz
*
Institute for General, Inorganic and Theoretical Chemistry, Innrain 52a, Leopold-Franzens-University Innsbruck, A-6020 Innsbruck, Austria. E-mail: hubert.huppertz@uibk.ac.at; Fax: +43(0)512507 2934; Tel: +43(0)512507 5100
First published on 21st September 2010
Abstract
Most of the syntheses in solid-state chemistry are performed at constant pressure of ∼1 atm (about 105 Pa) by the manipulation of the thermodynamic parameters temperature and composition, leading to a rich variety of compounds. In contrast, the additional variation of pressure has remained virtually unexplored, due to the relatively large costs of maintaining high-pressure conditions and the inevitably tiny sample volumes. In the last two decades, technical advances, developed for studying the properties of minerals, have found access into the preparative solid-state chemistry, opening up tremendously large areas to synthesize new materials.
 Hubert Huppertz | Hubert Huppertz studied chemistry at the University of Bayreuth (Germany), followed by a PhD thesis in the field of nitridosilicates in the group of Prof. W. Schnick. For habilitation, he changed to the Department of Chemistry of the Ludwig-Maximilians-Universität München (Germany), establishing a high-pressure laboratory for the syntheses of solid-state materials, followed by a habilitation thesis on new oxoborates through multianvil high-pressure/high-temperature syntheses. Recently, he took up a Chair in Inorganic Chemistry at the University of Innsbruck (Austria), where he is now going to explore the high-pressure/high-temperature chemistry and synthesis of new solid-state materials. |
Introduction
Over the past 70 years, the implementation of the parameter pressure into solid-state chemistry has led to remarkable enlarged synthetic possibilities. However, this field, studied from solid-state chemists with its spectrum of compounds and reactions so far, is really narrow, being just a beginning.1,2 Compared to the quite easily manageable parameters of temperature and composition, high-pressure conditions are technically much more demanding. The technical development of sophisticated high-pressure cells is in process, strongly depending on the improvements in materials chemistry (hard materials) and mechanical engineering.3–6 In this paper, the focus lies on the synthetic achievements made in our group by using a specific high-pressure equipment in different chemical systems.General remarks on high-pressure devices
From a historical point of view, gas-compressor systems, cold-seal vessels, and autoclaves were the first devices for reactions under enhanced pressure conditions. Bridgman's anvil apparatus and Bundy's recessed pair of anvils were steps on the way to the development of Hall's belt, which permitted experiments up to 10 GPa and simultaneously high temperatures up to 2000 °C. End-loaded and non-end-loaded piston-cylinder apparatus accompanied these developments with the advantage of higher experimental volumes and the disadvantage of a limited pressure range. The “belt” and the piston-cylinder apparatus were in fact the working horses for solid-state syntheses. Additionally, diamond anvil cells with built-in windows from diamond anvils started to become the most important device for in situ investigations up to the highest accessible pressures in the megabar regime. However, the latter technique is seriously affected due to the tiny sample volumes and therefore more suitable to investigate phase transformations of a known compound and less useful for the syntheses of new materials. Constructing a tetrahedral press in 1958, Hall7 launched the era of the multianvil technique, leading to many technical variations regarding the number of anvils, the assemblies positioned inside the anvils, and the compressing systems.8 The pressure generating capability of these devices strongly depends on the strength of the component material and the configuration of the specific apparatus. In general, multianvil devices can be used for solid-state syntheses in a pressure range up to 25 GPa and temperatures up to 3000 K with the advantage of a relatively large cell volume, which is highly important for synthesizing major quantities of metastable high-pressure materials.The modified Walker module
In comparison with the sophisticated multianvil systems, intending materials syntheses at high pressure and temperature in combination with large volumes at reasonable costs, the Walker module stands out from all other multianvil devices, meeting the mentioned requirements in a good way.9,10 Walker et al. developed this simplified multianvil module, based on a previous design of Ohtani et al.11 The modifications allowed a drastic reduction in hardware costs, avoided production obstacles, which occurred in current installations, and provided a considerably easier handling. A few years later, a modified Walker module was constructed by Frost et al. at the Bayerisches Geoinstitut (Bayreuth, Germany).12 Based on these modifications and the experiences of Frost's et al., we fabricated in cooperation with the company Voggenreiter (Mainleus, Germany) a Walker type module loadable up to a maximum of 1000 t(onne).5Fig. 1 illustrates the assemblage of a typical high-pressure assembly, including the cross section of the octahedral assembly, the joint-venture of the different parts of the modified Walker module, and the 1000 t press, which compresses the module to the demanded pressure. For heating, cylindrical resistance heaters from graphite or LaCrO3 surrounding the capsule material are used. Furthermore, the assembly can be prepared under argon in a glove-box, to handle air-sensitive compounds. A detailed description of a high-pressure experiment, using this module as the preparative basis of our work under high-pressure conditions, can be found in ref. 5. Starting with our work at the Department of Chemistry of the Ludwig-Maximilians-University Munich in 1999, the module has been used successfully in 15 other international laboratories. In the following, an overview about our achievements in new synthetic fields via high-pressure solid-state syntheses, based on the described technique, is given.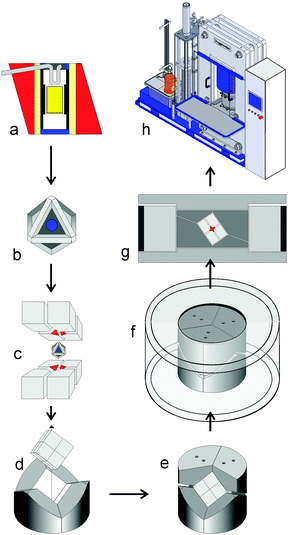 | ||
| Fig. 1 Schematic assemblage of a high-pressure experiment: (a) cross section of the octahedral pressure cell; (b) finished octahedron; (c) eight truncated tungsten carbide cubes to compress the octahedron; (d) three wedges incorporate the cube; (e) additional wedges from the top; (f) containment ring; (g) cross section of the module with pressure distribution plates; (h) uniaxial 1000 ton press. | ||
High-pressure chemistry
The majority of high-pressure science takes place in the geophysical area, where researchers try to identify the structural and physical properties of the minerals deep within our Earth.13 Furthermore, new areas have evolved in the last 20 years, e.g., the effect of pressure on living organisms or more general, the role of pressure in bioscience (food, pharmaceuticals, biomedical applications).14 Being chemists, we aim at the high-pressure/high-temperature synthesis of new and useful materials not yet available by other means. Due to the new technical possibilities, this is an emerging field in solid-state chemistry. At this point, one should briefly mention an important thermodynamic aspect in our research. As the new compounds are synthesized under high-pressure/high-temperature conditions, they are formed at the applied conditions of pressure and temperature, being thermodynamically stable at extreme conditions. Therefore, the reaction mixture is compressed up to the demanded pressure and afterwards heated to the specified reaction temperature. After the heating period, the sample is slowly decompressed to ambient-pressure conditions, recovered from the octahedron, and characterized by standard analytical methods. This procedure requires products, which can be decompressed to ambient-pressure conditions, where they are thermodynamically metastable, yet remain indefinitely kinetically stable. Thus, not every chemical system in solid-state chemistry is suitable for the synthesis of metastable high-pressure phases, because if the energy barrier between the high-pressure phase and the ambient-pressure phase is too low, the synthesized phase will retransform or decompose into thermodynamically stable compounds during the period of decompression. In the latter case, one has to investigate the specific system by in situ methods, where a continuous observation of the reaction process via e.g.X-ray diffraction (ESRF, Grenoble, France) is possible. In order to synthesize materials, chemical systems are preferable, in which the metastable compounds possess a high thermal stability under ambient pressure. From our group's research areas, the substance classes of borates, fluoride borates, and gallium oxonitrides are presented exemplarily, which fulfill these requirements, leading to a variety of new compounds.Borates
In nature, boron is always found covalently bonded to oxygen, forming an especially strong bond with it. Usually, boron compounds resemble those of other non-metals, notably silica. For example, B2O3 and SiO2 are similar in their acidic nature, and their melts readily dissolve metallic oxides to form borates and silicates that, in being cooled down, yield glasses declining to crystallize. Owing to additional trigonal BO3 groups next to BO4 tetrahedra, the great diversity of borate crystal structures may perhaps exceed even that of silicate structures. At the moment, more than 1000 borate crystal structures are listed in the Inorganic Crystal Structure Database ICSD.15 One of the main questions in oxoborate chemistry is a bonding model, which allows to calculate the ratio of trigonally to tetrahedrally coordinated boron atoms from the chemical formula.16 Several attempts were made to find an algorithm, but up to now, there is no general method to calculate the ratio in advance.17 Concerning most of the high-pressure phases, this model is unsuitable, because the fourfold coordination is favoured due to the pressure-coordination rule.18,19 The latter rule means that the structures respond to pressure by increasing coordination resulting in more densely packed structures. In contrast to normal-pressure investigations of borates, high-pressure investigations are rare. The major part of the results comes from geological research, investigating borate containing minerals. Following common belief, boron is mainly enriched in the continental crust of the earth due to its high volatility, where it forms borates in near-surface environments or borosilicates deep in the earth. Thus, considering the thickness of the normal continental crust, boron-bearing minerals should be restricted to the upper 30 kilometres of the globe, while mantle rocks are extremely poor in boron. Both conditions are caused and enhanced by the fact that, melting in the mantle, boron fractionates strongly into the liquid phase. Therefore, for a long time, studies on borates and borosilicates were limited to pressures below 1 GPa. The discovery of ultrahigh-pressure metamorphism of crustal rocks, occurring at depths of 100 km or more, led to considerable petrological and geochemical interest on the fate of boron in deeply subducted continental crusts.20 Several studies were performed by Werding and Schreyer, collecting data in the pressure range from 3 to 5 GPa.21 To investigate the synthesis and stability of boron bearing systems at elevated temperatures and pressures, they used hydrothermal bombs up to about 0.6 GPa, 650 to 800 °C, internally heated gas pressure vessels up to about 0.8 GPa, 1000 °C, and several types of piston-cylinder apparatus up to 6 GPa, 1100 °C. Only a few experiments were performed at higher pressures (>6 GPa) with a multianvil apparatus at the Bayerisches Geoinstitut, Bayreuth (Germany). So, less is known about the high-pressure behaviour of oxoborates in a pressure range larger than 6 GPa.With the Walker-type multianvil apparatus, it is now possible to perform experiments with pressures up to 25 GPa, considerably extending the experimental area and gaining new insights into the structural and synthetic possibilities of oxoborate chemistry. In the following, some examples from our research are presented, which can be devided into three sections: (1) the syntheses of new polymorphs of already existing ambient-pressure phases; (2) the syntheses of compounds with new compositions not attainable under ambient-pressure conditions; (3) the access to crystalline phases of borates, forming glasses under ambient-pressure conditions.
Syntheses of new polymorphs
During our high-pressure/high-temperature syntheses of new polymorphs, we discovered several new high-pressure polymorphs of known ambient-pressure oxoborates, e.g., β-MB4O7 (M = Ca, Zn, Hg),22–24χ-REBO3 (RE = Dy,Er),25 and ν-DyBO3.26 A special attention was given to the rare-earth oxoborates, which already show polymorphism at normal pressure conditions. A closer look at the meta-oxoborates RE(BO2)3 revealed the well-characterized monoclinic phases α-RE(BO2)3 (RE = La–Nd, Sm–Tb)27–29 and the orthorhombic phases β-RE(BO2)3 (RE = Tb), the latter solved by Nikelski and Schleid in 2003 for the first time.30Fig. 2 (top) shows the crystal structure of the α-type modification. These compounds are characterized by chains of tetrahedral BO4 and triangular BO3 units, running along the c axis and built up on the regular succession of BO4 tetrahedra, which share four corners with monodentate BO3 triangles.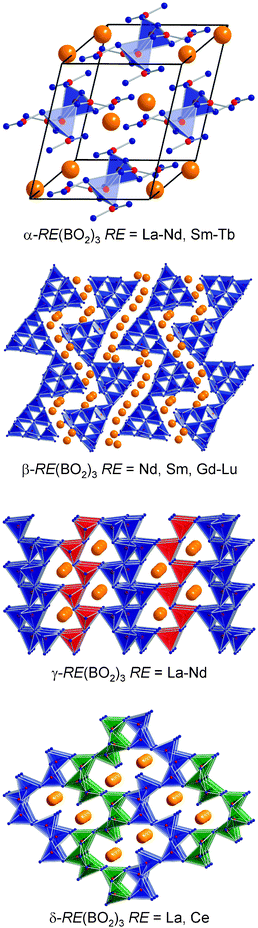 | ||
| Fig. 2 Crystal structures of the four structure types of the rare-earth oxoborates RE(BO2)3. | ||
The crystal structure of the meta-oxoborates β-RE(BO2)3 (RE = Tb–Lu) consists of strongly corrugated layers, composed of corner-shared BO4 tetrahedra (Fig. 2, second from the top). Besides the bridging oxygen atoms (O[2]), the structure contains also threefold coordinated oxygen atoms O[3]. The further research with smaller rare-earth metal ions led to the result that only the dysprosium compound was attainable under normal-pressure conditions. For the synthesis of the remainder β-RE(BO2)3 series with RE = Ho–Lu using the corresponding rare-earth oxide and boron oxide as starting materials, high-pressure/high-temperature conditions of 7.5 GPa and 1000 °C were necessary.31
Due to the fact that the monoclinic phases α-RE(BO2)3 were built up from trigonal BO3 and tetrahedral BO4 groups, we assumed a possible transformation into a new modification by applying high-pressure conditions. This assumption was fulfilled by the synthesis of the orthorhombic phases γ-RE(BO2)3 (RE = La–Nd) at conditions of 7.5 GPa and 1000 °C.32,33 As expected for a high-pressure phase in oxoborate chemistry, the structure-type consists purely of corner-sharing BO4 tetrahedra, forming a three-dimensional network (Fig. 2, third from the top). In detail, the crystal structure contains layers of BO4 tetrahedra, further connected via zigzag chains of BO4 tetrahedra. Here, too, threefold coordinated oxygen atoms, O[3], are part of the layers. Naturally, the difference between normal-pressure and high-pressure of 7.5 GPa is remarkable. For example, the calcium borate CaB2O4 exhibits three phase transformations up to a pressure of 4 GPa. Therefore, we analyzed the pressure range below 7.5 GPa and discovered a fourth structure type designated as δ-RE(BO2)3 for RE = La and Ce at 5.5 GPa and 3.5 GPa, respectively.28,34 The crystal structure of these δ-phases is built up exclusively from BO4 tetrahedra, which are linked via common corners to form a network. Fig. 2 (bottom) gives a view of the crystal structure along [100], showing large ten-membered rings occupied by the rare-earth ions, and small, empty four-membered rings.
This example from the rare-earth oxoborates shows remarkably, how pressure can be used to enlarge structure types of known compounds. Additional structure types at higher pressures can be imagined, as the former mentioned high-pressure compounds were all synthesized below 8 GPa.
A second example of a new high-pressure polymorph comes from nonlinear optical (NLO) materials, which are used to generate new lasers with frequencies, that cannot be obtained directly. Since the discovery of the first effective borate material β-BaB2O4 (BBO) being suitable for frequency conversion, the potential of a large variety of borates has been investigated under ambient pressure.35 The most prominent compound in this system is bismuth triborate (BiB3O6, BIBO) because of its exceptional nonlinear optical properties, which has excited the materials community.36–39 In 2005, Liet al. synthesized two new polymorphs of BiB3O6.40 For the sake of clarity, the authors renamed the original bismuth triborate as α-BiB3O6 and the two new modifications as β- and γ-BiB3O6. In contrast to the α phase, which crystallizes in the non-centrosymmetric, monoclinic space groupC2, these modifications crystallize in the centrosymmetric space groupP21/n. Under high-pressure conditions of 5.5 GPa and a temperature of 820 °C, we successfully synthesized a fourth modification δ-BiB3O6 (from a stoichiometric mixture of Bi2O3 and B2O3), which has got again a non-centrosymmetric structure (space group: Pca21).41 As the interatomic spacing is reduced under pressure, one could expect a higher symmetry for the high-pressure phase δ-BiB3O6. However, the center of inversion is lacking in the space groupPca21, prompting to investigate the nonlinear optical properties. Fig. 3 gives a view of the four different modifications of BiB3O6.
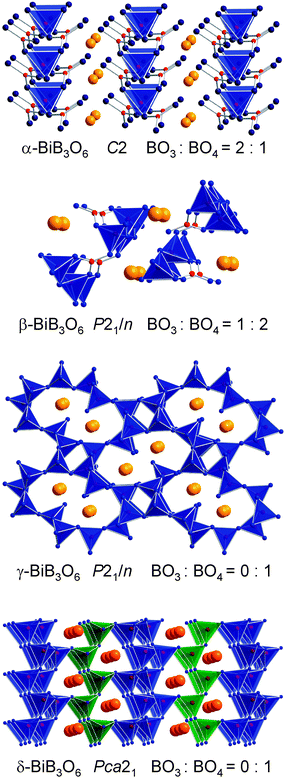 | ||
| Fig. 3 The four different polymorphs of BiB3O6. | ||
A comparison between α-, β-, and γ-BiB3O6 shows an increasing density (α: 5.033; β: 5.411; γ: 6.177 g cm−3), running parallel with a transformation of BO3 into BO4 groups. In the ambient-pressure modification α-BiB3O6, the ratio of BO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶BO4 is 2∶1, and this ratio changes to 1∶2 in β-BiB3O6. The structure of γ-BiB3O6 shows only BO4 tetrahedra. As expected through the high-pressure synthesis, the new phase δ-BiB3O6 exhibits a denser structure (6.378 g cm−3) than the γ modification, prepared under ambient-pressure conditions. Due to the fact that the coordination of the oxygen atoms is identical in both structures (fraction of O[3]: 1/6), the bismuth coordination must be responsible for the higher density. In fact, δ-BiB3O6 has sevenfold-coordinated Bi3+ ions in contrast to sixfold-coordinated Bi3+ ions in γ-BiB3O6. A powder sample of δ-BiB3O6 was subjected to a qualitative powder second-harmonic-generation (SHG) measurement. The intensities of the second harmonics generated by δ-BiB3O6 and by the reference compound KDP, were nearly equal, thus corroborating the non-centrosymmetry of δ-BiB3O6 and indicating a considerable SHG effect of the compound. Interestingly, the structure types of δ- and γ-BiB3O6 are isostructural to the former mentioned compounds γ-RE(BO2)3 (RE = La–Nd) and δ-RE(BO2)3 (RE = La, Ce), respectively. A specific difference lies in the coordination sphere of the metal cations, which is caused by the sterically active lone pair, localized at each Bi3+ center.
∶BO4 is 2∶1, and this ratio changes to 1∶2 in β-BiB3O6. The structure of γ-BiB3O6 shows only BO4 tetrahedra. As expected through the high-pressure synthesis, the new phase δ-BiB3O6 exhibits a denser structure (6.378 g cm−3) than the γ modification, prepared under ambient-pressure conditions. Due to the fact that the coordination of the oxygen atoms is identical in both structures (fraction of O[3]: 1/6), the bismuth coordination must be responsible for the higher density. In fact, δ-BiB3O6 has sevenfold-coordinated Bi3+ ions in contrast to sixfold-coordinated Bi3+ ions in γ-BiB3O6. A powder sample of δ-BiB3O6 was subjected to a qualitative powder second-harmonic-generation (SHG) measurement. The intensities of the second harmonics generated by δ-BiB3O6 and by the reference compound KDP, were nearly equal, thus corroborating the non-centrosymmetry of δ-BiB3O6 and indicating a considerable SHG effect of the compound. Interestingly, the structure types of δ- and γ-BiB3O6 are isostructural to the former mentioned compounds γ-RE(BO2)3 (RE = La–Nd) and δ-RE(BO2)3 (RE = La, Ce), respectively. A specific difference lies in the coordination sphere of the metal cations, which is caused by the sterically active lone pair, localized at each Bi3+ center.
Thus, the systematic investigation of the phase diagrams of known compounds, taking into account the parameter pressure, might give access to useful nonlinear optical materials. Therefore, we intend a systematic investigation of new noncentrosymmetric borates synthesized under high-pressure conditions. The question is, whether sufficient amounts of a specific compound can be synthesized for an industrial application? The artificial synthesis of diamond under extreme conditions of pressure and temperature indicates impressively that compounds in the scale of tons can be synthesized under high-pressure conditions, if they are of interest and the pressure does not exceed a value of 6–8 GPa. A second point is the access to small single crystals of the high-pressure phase, which could serve as a seed for crystal growth of the compound in question under ambient-pressure conditions. Third, the high-pressure synthesis can lead to low-temperature modifications, because often the high-pressure modification of a known compound corresponds to a not yet discovered low-temperature form at ambient-pressure conditions. In the case of δ-BiB3O6, Cong et al. were recently able to show that the high-pressure phase corresponds to the low-temperature form, which can be realized at ambient-pressure conditions via a phase transformation starting from β-BiB3O6 after 100 h annealing at 650–670 °C.42
The syntheses of new polymorphs of existing compositions is a highly interesting topic, which is often performed in diamond-anvil cells with the advantage of the in situdetection of phase transformations. A much more demanding challenge is the synthesis of new compositions, whose formation is indispensably based on high-pressure conditions during the syntheses—the topic of the next section.
Syntheses of compounds with new compositions
One of the most interesting topics in high-pressure solid-state chemistry is the synthesis of compounds with compositions, which are completely unknown at ambient pressure. At the beginning of our research in 1999, trivalent rare-earth oxoborates of the composition REBO3, REB3O6, “RE3BO6”, RE26O27(BO3)8, RE17.33(BO3)4(B2O5)2O16, and RE8.66(BO3)2(B2O5)O8 were found in the literature. Table 1 gives a survey of all known compositions (to the best of our knowledge) of ternary rare-earth oxoborates with the specified trivalent rare-earth cations, ordered by the ratio RE2O3∶B2O3. Partly, several polymorphs of these compositions could be obtained at higher temperatures and pressures and a detailed reference list for all compounds can be found in ref. 43. Later on, the compositions REB5O9 and RE4B14O27 were discovered under normal-pressure conditions by Liet al. and Nikelski et al., respectively.44–46
∶B2O3 (new compositions are shown in italics)
| Composition | RE |
RE2O3:B2O3 |
RE:B |
|---|---|---|---|
| α-REB5O9 | Sm–Er | 1:5 |
0.2 |
| β-REB5O9 | La, Ce | 1:5 |
0.2 |
| RE4B14O27 | La | 1:3.5 |
0.285 |
| α-REB3O6 | La–Nd, Sm–Tb | 1:3 |
0.33 |
| β-REB3O6 | Nd, Sm, Gd–Lu | 1:3 |
0.33 |
| γ-REB3O6 | La–Nd | 1:3 |
0.33 |
| δ-REB3O6 | La, Ce | 1:3 |
0.33 |
| RE4B10O21 | Pr |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :2.5 :2.5
|
0.4 |
| α-RE2B4O9 | Sm–Ho |
1:2
|
0.5 |
| β-RE2B4O9 | Gd , Dy |
1:2
|
0.5 |
| RE3B5O12 | Er–Lu |
3:5
|
0.6 |
| RE4B6O15 | Dy , Ho |
2:3
|
0.67 |
| π-REBO3 | Y, Nd, Sm–Lu | 1:1 |
1 |
| μ-REBO3 | Y, Sm–Gd, Dy–Lu | 1:1 |
1 |
| λ-REBO3 | La–Nd, Sm, Eu | 1:1 |
1 |
| β-REBO3 | Sc, Er–Lu | 1:1 |
1 |
| ν-REBO3 | Ce–Nd, Sm–Dy | 1:1 |
1 |
| χ-REBO3 | Dy–Er | 1:1 |
1 |
| H-REBO3 | La, Ce, Nd | 1:1 |
1 |
| RE8.66O8(BO3)2(B2O5) | Ho | 8.66:4 |
2.165 |
| RE17.33O16(BO3)4(B2O5)2 | Y, Gd | ∼8.7:4 |
2.175 |
| RE3BO6 | Y, La, Pr–Lu | 3:1 |
3 |
| RE26O27(BO3)8 | La, Nd | 13:4 |
3.25 |
| RE31O27(BO3)3(BO4)6 | Ho |
31:9
|
3.44 |
Applying high-pressure/high-temperature conditions on stoichiometric mixtures of rare-earth oxides and boron oxide, we were able to synthesize five new compositions in this system, namely, RE3B5O12,47RE4B10O21,48,49RE31O27(BO3)3(BO4)6,43RE4B6O15,50,51 and RE2B4O952–56 (in italics in Table 1). These compounds possess a remarkable metastability up to 600–800 °C before they decompose into the corresponding ortho-borates or meta-borates under ambient-pressure conditions. For example, successive heating of Yb3B5O12 from the series RE3B5O12 (RE = Er–Lu) in the range of 800–900 °C led to a decomposition into a mixture of μ-YbBO3 and π-YbBO3. The compounds crystallize into a novel structure type, being homeotypic with the beryllosilicate mineral semenovite ((Fe2+, Mn, Zn, Ti)RE2Na0−2(Ca, Na)8(Si, Be)20(O, OH, F)48). Thus, the compounds RE3B5O12 (RE = Er–Lu) represent an outstanding example of complex silicate structures in the field of borates using high pressure.
With the high-pressure synthesis of RE4B10O21 (RE = Pr) at a relatively mild pressure of 3.5 GPa, we obtained a rare-earth oxoborate structure of BO3 and BO4 groups in the ratio 1:4, which are linked to a highly connected network in accordance with the relatively high amount of boron in the formula (RE2O3:B2O3 = 1:2.5).
On the other hand, we were able to synthesize the compound RE31O27(BO3)3(BO4)6 (RE = Ho) with the lowest percentage of boron in a trivalent rare-earth oxoborate up to now.43Fig. 4 shows the structure of Ho31O27(BO3)3(BO4)6, displaying isolated BO4 tetrahedra as dark hatched polyhedra and BO3 units in form of ball-and-stick models, embedded in a complex holmium oxide network.
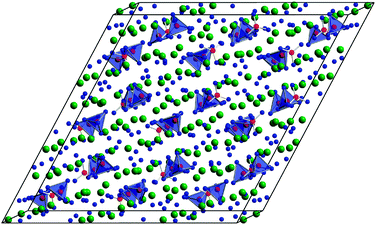 | ||
| Fig. 4 Crystal structure of Ho31O27(BO3)3(BO4)6. (BO3 groups are shown in form of a ball-and-stick model; BO4 tetrahedra are visualized through polyhedra; large spheres: Ho3+, small spheres: O2−). | ||
RE31O27(BO3)3(BO4)6 (RE = Ho) represents the most metal rich rare-earth oxoborate known up to now. Based on our experience, we would have expected exclusively BO4 tetrahedra for a compound, which was synthesized under the extreme pressure conditions of 11.5 GPa (pressure coordination rule).18,19 Nevertheless, Ho31O27(BO3)3(BO4)6 also contains trigonal-planar coordinated boron atoms, which are exceptional for a material synthesized under such conditions. In the past, all oxoborate compounds synthesized at pressures exceeding 8 GPa revealed exclusively BO4 tetrahedra. All the more it is astonishing that the BO3 group is stable in the structure of Ho31O27(BO3)3(BO4)6, especially in the plethora of oxygen atoms.
In this context, the general influence of increased pressure on the structure of new oxoborates attracts our intention. It was already mentioned that the coordination number of the boron atoms can be enhanced from three to four. Additionally, the metal cations and the oxygen anions (O[2] → O[3]) often reveal higher coordination numbers in comparison with compounds under normal pressure conditions. However, the most striking discovery was the structural motif of edge-sharing BO4 tetrahedra ([B2O6]6− units), which was observed for the first time in 2002 in the rare-earth borate Dy4B6O15 and two years later in the isotypic holmium phase Ho4B6O15.50,51Fig. 5 gives a view of the corresponding crystal structures.
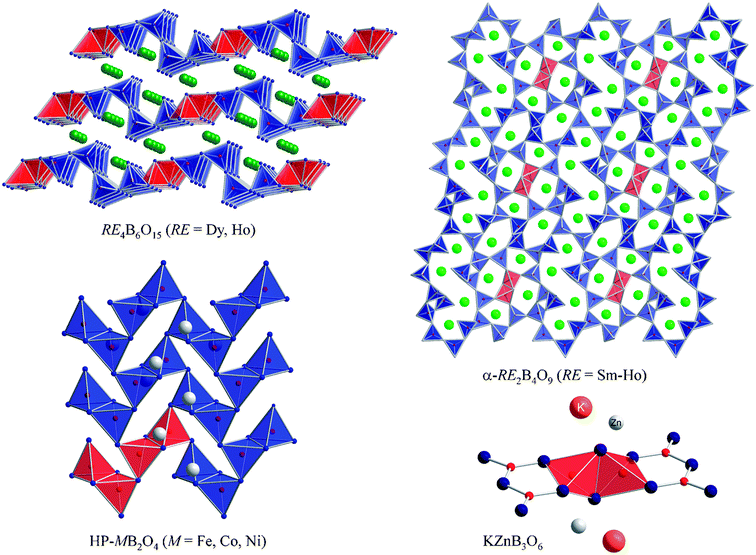 | ||
| Fig. 5 Crystal structures of the four known structure types in the chemistry of oxoborates, which exhibit the structural motif of edge-sharing BO4 tetrahedra (partially marked as red polyhedra). Except for KZnB3O6, these compounds were synthesized under high-pressure conditions. | ||
A second series of borates, having edge-sharing BO4 tetrahedra in a different structure type, could be synthesized with the composition α-RE2B4O9 (RE = Sm–Ho).52–54 In these examples, only a fraction of 1/3 (RE4B6O15 (RE = Dy, Ho)) or 1/10 (α-RE2B4O9 (RE = Sm–Ho)) of the BO4 tetrahedra bridge to a second tetrahedron via a common edge. The corresponding polymorphs β-RE2B4O9 (RE = Dy, Gd) did not show BO4 tetrahedra with common edges.55,56 The latest high-pressure experiments with transition-metal borates yielded a third structure type with edge-sharing tetrahedra. The special feature of the compound HP-NiB2O4 is that in contrast to the first two compounds, all tetrahedra are linked to each other via one common edge and two common corners.57 Meanwhile, it was possible to prepare the isotypic structures β-FeB2O458 and HP-CoB2O4.59 To our astonishment, the structural feature of edge-sharing BO4 tetrahedra was recently found in the compound KZnB3O6, which was synthesized under ambient-pressure conditions.60,61 This result means that the structural motif of edge-sharing BO4 tetrahedra is no longer a domain of high-pressure chemistry, but still favoured under these conditions, as there exist eleven different compounds with three structure types prepared under high-pressure and only one compound synthesized under ambient-pressure conditions. Fig. 5 summarizes all four structure types known up to now, in which the structural motif of edge-sharing BO4 tetrahedra was observed.
Additionally to the syntheses of new compounds with fascinating structural features, high-pressure gives us access into a variety of systems, which are normally glass formers. The next section will give a view of the power of high-pressure in glass forming systems.
Pressure induced crystallization of glassy borates
Under ambient-pressure conditions, oxoborates are pronounced glass formers.62 For example, the syntheses of crystalline forms of the three ambient-pressure modifications of bismuth triborate from the melt is hindered due to a high viscosity of the melt, making it difficult to obtain a crystalline compound. For the synthesis of δ-BiB3O6, high-pressure was applied, leading to a highly crystalline sample (vide supra). Two factors support the crystallization: the closed sample capsule prevents the evaporation of boron oxide, thus keeping the composition constant, and the high temperature assures a high mobility of the specific ions, which enables the reconstructive formation of a denser phase of bismuth triborate. The importance of an adequate temperature to overcome the kinetic barrier for the transformation into a high-pressure phase is underlined by the fact that we never observed a pressure induced amorphization of the materials. In most of the syntheses of new high-pressure borates we believe that the syntheses and crystallization occurs from the melt. This is confirmed by the observation that fast quenching after the heating period can lead to glasses.In this context, we had a look into the ternary system Sn–B–O as a simplified variant of the tin-based amorphous composite oxides (TCO), materials in use as negative electrodes of lithium-ion rechargeable batteries. Several investigations were performed in this system and because all compounds in this area were glasses, the synthesis of crystalline approximands for a more detailed structural investigation would be favourable. In fact, the use of high-pressure/high-temperature of 7.5 GPa and 1100 °C led to the synthesis of the first crystalline tin borate β-SnB4O7.63Fig. 6 gives a view of the crystal structure of β-SnB4O7, which is purely built up of corner-sharing BO4 tetrahedra, being isotypic to the known high-pressure phases β-CaB4O722 and β-HgB4O7,24 as well as to the ambient-pressure phases SrB4O7,64,65PbB4O7,66 and EuB4O7.67
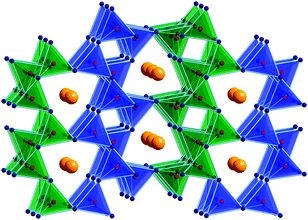 | ||
| Fig. 6 Crystal structure of β-SnB4O7. | ||
Generally, we would expect that the nucleation and growth of a crystalline phase from the amorphous “SnB4O7” with increasing pressure gets impeded by a suppressed ionic mobility at higher pressures. However, these observations suggest that the reconstructive formation of the more densely packed compound β-SnB4O7 under pressure is kinetically not restricted at a temperature of 1100 °C.
In a further step, we had a look at the ternary systems Hf–B–O and Zr–B–O, where no defined crystalline materials could be obtained via conventional synthetic routes until then. The experiments in the ternary system Hf–B–O were successful with the synthesis of the first hafnium diborate β-HfB2O5 starting from a stoichiometric mixture of HfO2 and B2O3.68 Due to the fact that this compound is a high-pressure phase, we labelled it with the Greek character “β”. Later on, it became also possible to synthesize the isotypic zirconium phase β-ZrB2O5 under similar conditions.69 Attempts to synthesize the ambient-pressure hafnium diborate and zirconium diborate “α-MB2O5 (M = Zr, Hf)” are ongoing. Fig. 7 gives a view of the crystal structure of β-MB2O5 (M = Zr, Hf), showing layers of BO4 tetrahedra and M4+ (M = Zr, Hf) ions.
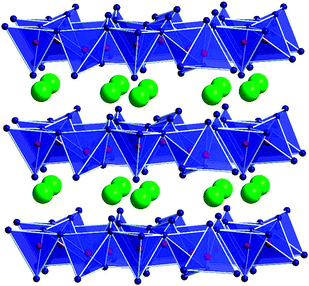 | ||
| Fig. 7 Crystal structure of β-MB2O5 (M = Zr, Hf). | ||
The arrangement of the BO4 tetrahedra in β-MB2O5 (M = Zr, Hf), with four- and eight-membered rings, shows that the topology of the tetrahedra is identical to the arrangement in silicate minerals of the gadolinite group.70 Several important minerals and synthetic compounds belong to the gadolinite group (space groupP21/c), which can be represented by the general chemical formula A2Z2XSi2O8(O,OH)2. In this context, the compounds β-MB2O5 (M = Zr, Hf) can be considered as the simplest structural variant of all compounds belonging to the complex gadolinite family. For a more detailed discussion of the interesting structural relationships, the reader is referred to ref. 68.
The latter examples show impressively that pressure favours the formation of crystalline oxoborates in contrast to normal-pressure investigations, which often lead to amorphous compounds (glasses). In the future, we hope to have access to a large variety of other systems, of which no compounds exist until now.
Fluoride borates
Considering the extensive research on oxoborates under high-pressure/high-temperature conditions in our group in the last decade, it was tempting to extend our investigations into fluoride borates. These compounds, especially “fluoroborate” glasses, have been object of extensive research.71–73 The doping with rare-earth element cations leads to interesting luminescence, fluorescence, and even dielectric properties of the glasses.74–77 It is known that “fluoroborate” glasses, as well as borate glasses, are UV-transmitting; the addition of fluorine enlarges the optical gap.78,79 Crystalline fluorido and fluoride borates are not that well studied, excluding natural minerals, with more than two cations. Under extreme synthetic conditions, the number and variety of oxoborates could be greatly extended, revealing fascinating structures and attractive material properties. Due to their structural similarities to fluoride borates, it is more than likely, that numerous new compounds and crystal structures can be obtained here under pressure. Among the existing fluoride borates, there are mainly trigonal BO3 groups, mostly connected via common corners. Consequently, the field of fluoride borates has an enormous potential for high-pressure investigations. The connection of existing building blocks or the increase of coordination numbers could lead to a tremendous number of higher condensed structures.In a first approach, it was possible to synthesize a new series of fluoride borates with the composition RE5(BO3)2F9 (RE = Er, Tm, Yb) (Fig. 8) starting from stoichiometric mixtures of RE2O3, REF3, and B2O3.80–82
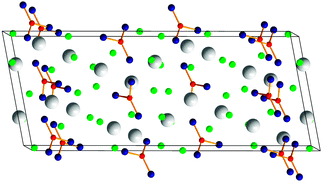 | ||
| Fig. 8 Crystal structure of the fluoride borates RE5(BO3)2F9 (RE = Er, Tm, Yb); BO3 groups are shown in the form of a ball-and-stick model; large spheres: RE3+, small spheres: F−. | ||
These rare-earth fluoride borates exclusively consist of isolated trigonal BO3 groups, rare-earth cations, and fluoride anions, closely related to the compounds RE3(BO3)2F3 (RE = Sm, Eu, Gd)83,84 and Gd2(BO3)F3.85 In fact, all three structure types can be described by different sequences of layers with the formal composition “REBO3” and “REF3”.
Since the compounds RE5(BO3)2F9 (RE = Er, Tm, Yb) contain only trigonal BO3 groups, it was attractive to synthesize a fluoride borate with BO4 groups by higher pressure. Consequently, we were able to synthesize Gd4B4O11F2, in which BO3 groups and BO4 tetrahedra were found, connected via common corners (Fig. 9).86
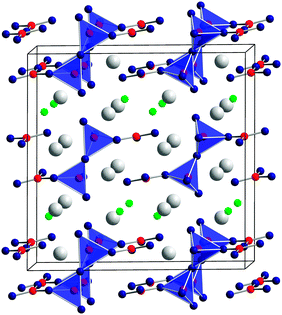 | ||
| Fig. 9 Crystal structure of Gd4B4O11F2, showing the fundamental building block units, consisting of BO3 groups and BO4 tetrahedra. Large spheres: Gd3+, small spheres: F−. | ||
A much more complicated structure is found for the lanthanum fluoride borate La4B4O11F2, which shows the same formula but a different rare-earth cation.87 The crystal structure is fairly complicated with a large unit cell built up from BO4 tetrahedra interconnected with two BO3 groupsvia common vertices, B2O5-pyroborate units, and isolated BO3 groups. Fig. 10 gives a view of the crystal structure of La4B4O11F2.
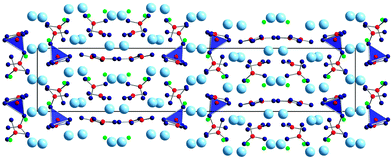 | ||
| Fig. 10 Crystal structure of La4B4O11F2, consisting of La3+ and F− ions, BO3 groups and BO4 tetrahedra. | ||
The b lattice parameter is about five times longer than the other cell edges. Looking at the structure along c, a wave-like modulation of the cations and the B–O-polyhedra with a formal wavelength λ = b was observed. In contrast to the well known Vernier phases,88–91 no separation into mismatching substructures was found in La4B4O11F2. In analogy to the meta-oxoborates, continuing experiments on the formation range of the compounds RE4B4O11F2 will be undertaken. It will be of great interest to see, which crystal structure is favoured by the rare-earth ions of intermediate size and whether it will be possible to obtain both structure types as polymorphs for one and the same cation.
In principle, most of the working areas in solid-state chemistry can be extended into the regime of high-pressure. This often results in interesting collaborations, e.g. in the field of gallium oxonitrides.
Gallium oxonitrides
In contrast to the well-known phase diagram of the binary phase system Al2O3 and AlN,92 the knowledge of the respective phases in the system Ga2O3 and GaN was fragmentary for a long time. The first compound in the system Ga–O–N with the composition Ga1−x/3□x/3N1−xOx with x ∼ 0.1 (□ = vacancy) was mentioned by Verdier and Marchand.93 Further publications and patents on gallium oxonitrides as passivation films and insulating layers were presented by Shiota and Nishizawa, in which the usefulness and effectiveness of gallium oxonitride as an optical and electrical material was pointed out.94–96 In 2005, McMillan's group in London and our joint cooperation group published the high-pressure/high-temperature synthesis of spinel-type gallium oxonitride phases for the first time.97,98 While we used an amorphous/nanocrystalline gallium oxonitride ceramic, obtained by the pyrolysis of a precursor [Ga(OtBu)2NMe2]2, with a mean N/O ratio of 0.86 as starting material, Soignard et al. used a mixture of α/β-Ga2O3 and w-GaN in a molar ratio of 1:1. Later on, we systematically investigated the stability field of this spinel phase by reactions between hexagonal GaN and monoclinic β-Ga2O3 with different molar ratios under specific high-pressure/high-temperature conditions.99 Therewith, initial conditions of 2.5–11.5 GPa at temperatures between 1000–1300 °C could be stated as leading to the formation of cubic, spinel-type gallium oxonitride phases.
From one of these experiments, (GaN:Ga2O3 = 9:1; 5 GPa, 1250 °C), the isolation of single crystals of spinel-type gallium oxonitride became possible for the first time.100 The combination of energy dispersive X-ray spectroscopy (EDS) with electron energy-loss spectroscopy (EELS) allowed the quantification of nitrogen and oxygen for the structural refinement of the single-crystal data (N/O = 0.25 ± 0.06). The observation of a site occupation deficiency of the octahedral gallium site led to the composition Ga2.79□0.21(O3.05N0.76□0.19). In the literature on oxonitrides, crystal defects in spinel-type materials are handled with different models, mainly by the assumption of a constant anion model.101–103 The results of the single-crystal structure determination indicate that this model is doubtful, and one should take into account a model with both cation and anion vacancies. A more detailed discussion of the single-crystal refinement and the consequences for the occupation of the crystallographic sites can be found in ref. 100.
Furthermore, we performed in situdiamond anvil cell (DAC) investigations, using the end members w-GaN and β-Ga2O3 in a first route and an amorphous gallium oxonitride ceramic in a second one.104 Both approaches led to remarkable new insights into the formation tendencies of gallium oxonitride spinel-type compounds.
Conclusions
In the past, solid-state chemistry has predominantly been performed by the variation of the thermodynamic parameters “composition” and “temperature”. The universal usage of the thermodynamically equal parameter “pressure” during the syntheses, which is routinely possible in our group, opens up tremendous possibilities for the synthesis and manipulation of new materials. This article introduces recent new developments in the field of borates, fluoride borates, and gallium oxonitrides. Furthermore, our group is active in the fields of intermetallic compounds, cuprates, chromates, phosphates, germanates, arsenates and oxides, revealing fascinating new areas of solid-state chemistry.Acknowledgements
The author gratefully acknowledges the continuous support of this work by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.Notes and references
- P. F. McMillan, Chem. Commun., 2003, 919 RSC.
- W. Grochala, R. Hoffmann, J. Feng and N. W. Ashcroft, Angew. Chem., Int. Ed., 2007, 46, 3620 CrossRef CAS.
- D. C. Rubie, Phase Transitions, 1999, 68, 431 CrossRef CAS.
- R. Boehler, Rev. Geophys., 2000, 38, 221 CrossRef CAS.
- H. Huppertz, Z. Kristallogr., 2004, 219, 330 CrossRef CAS.
- D. J. Frost, B. T. Poe, R. G. Tronnes, C. Liebske, A. Duba and D. C. Rubie, Phys. Earth Planet. Inter., 2004, 143–144, 507 CrossRef CAS.
- T. Hall, Rev. Sci. Instrum., 1958, 29, 267 CrossRef.
- A. Onodera, High Temp.–High Pressures, 1987, 19, 579 CAS.
- D. Walker, M. A. Carpenter and C. M. Hitch, Am. Mineral., 1990, 75, 1020.
- D. Walker, Am. Mineral., 1991, 76, 1092.
- E. I. Ohtani, T. Irifune, W. O. Hibberson and A. E. Ringwood, High Temp.–High Pressures, 1987, 19, 523.
- D. J. Frost, personal communication.
- Ultrahigh-Pressure Mineralogy: Physics and Chemistry of the Earth's Deep Interior, in Rev. Mineral., ed. R. J. Hemley, Mineralogical Society of America, Washington, 1998, vol. 37 Search PubMed.
- N. Rivalain, J. Roquain and G. Demazeau, Biotechnol. Adv., 2010 DOI:10.1016/j.biotechadv.2010.04.001.
- Inorganic Crystal Structure Data Base—ICSD, Fachinformationszentrum (FIZ) Karlsruhe, Karlsruhe, 2010, http://icsd.fiz-karlsruhe.de/.
- J. Krogh-Moe, J. Non-Cryst. Solids, 1969, 1, 269 CrossRef CAS.
- E. Parthé, Z. Kristallogr., 2002, 217, 179 CrossRef CAS.
- A. Neuhaus, Chimia, 1964, 18, 93 CAS.
- C. T. Prewitt and R. T. Downs, Rev. Mineral., 1998, 37, 283 Search PubMed.
- C. Chopin, Contrib. Mineral. Petrol., 1984, 86, 107 CrossRef CAS.
- G. Werding and W. Schreyer, in Boron, Mineralogy, Petrology and Geochemistry, ed. E. S. Grew and L. M. Anovitz, Mineralogical Society of America, Washington, 1996, vol. 33 Search PubMed.
- H. Huppertz, Z. Naturforsch., Teil B, 2003, 58, 257 CAS.
- H. Huppertz and G. Heymann, Solid State Sci., 2003, 5, 281 CrossRef CAS.
- H. Emme, M. Weil and H. Huppertz, Z. Naturforsch., Teil B, 2005, 60, 815 CAS.
- H. Huppertz, B. von der Eltz, R.-D. Hoffmann and H. Piotrowski, J. Solid State Chem., 2002, 166, 203 CrossRef CAS.
- H. Emme and H. Huppertz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, i117 CrossRef.
- J.-S. Ysker and W. Hoffmann, Naturwissenschaften, 1970, 57, 129 CrossRef CAS.
- G. Heymann, T. Soltner and H. Huppertz, Solid State Sci., 2006, 8, 821 CrossRef CAS.
- G. K. Abdullaev, K. S. Mamedov and G. G. Dzhfarov, Sov. Phys. Crystallogr., 1981, 26, 473 Search PubMed.
- T. Nikelski and T. Schleid, Z. Anorg. Allg. Chem., 2003, 629, 1017 CrossRef CAS.
- H. Emme, T. Nikelski, T. Schleid, R. Pöttgen, M. H. Möller and H. Huppertz, Z. Naturforsch., Teil B, 2004, 59, 202 CAS.
- H. Emme, C. Despotopoulou and H. Huppertz, Z. Anorg. Allg. Chem., 2004, 630, 1717 CrossRef.
- H. Emme, C. Despotopoulou and H. Huppertz, Z. Anorg. Allg. Chem., 2004, 630, 2450 CrossRef CAS.
- A. Haberer, G. Heymann and H. Huppertz, Z. Naturforsch., Teil B, 2007, 62, 759 CAS.
- P. Becker, Adv. Mater., 1998, 10, 979 CrossRef CAS.
- J. Liebertz, Z. Kristallogr., 1982, 158, 319 CAS.
- R. Fröhlich, L. Bohatý and J. Liebertz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 343 CrossRef.
- P. Becker, J. Liebertz and L. Bohatý, J. Cryst. Growth, 1999, 203, 149 CrossRef CAS.
- H. Hellwig, J. Liebertz and L. Bohatý, Solid State Commun., 1998, 109, 249 CrossRef.
- L. Li, G. Li, Y. Wang, F. Liao and J. Lin, Inorg. Chem., 2005, 44, 8243 CrossRef CAS.
- J. S. Knyrim, P. Becker, D. Johrendt and H. Huppertz, Angew. Chem., Int. Ed., 2006, 45, 8239 CrossRef CAS.
- R. Cong, J. Zhu, Y. Wang, T. Yang, F. Liao, C. Jin and J. Lin, CrystEngComm, 2009, 11, 1971 RSC.
- S. A. Hering, A. Haberer, R. Kaindl and H. Huppertz, Solid State Sci., 2010, 12 DOI:10.1016/j.solidstatesciences.2010.08.016.
- L. Li, P. Lu, Y. Wang, X. Jin, G. Li, Y. Wang, L. You and J. Lin, Chem. Mater., 2002, 14, 4963 CrossRef CAS.
- L. Li, X. Jin, G. Li, Y. Wang, F. Liao, G. Yao and J. Lin, Chem. Mater., 2003, 15, 2253 CrossRef CAS.
- T. Nikelski, M. C. Schäfer and T. Schleid, Z. Anorg. Allg. Chem., 2008, 634, 49 CrossRef CAS.
- H. Emme, M. Valldor, R. Pöttgen and H. Huppertz, Chem. Mater., 2005, 17, 2707 CrossRef CAS.
- A. Haberer, G. Heymann and H. Huppertz, Z. Anorg. Allg. Chem., 2006, 632, 2079 CrossRef.
- A. Haberer, G. Heymann and H. Huppertz, J. Solid State Chem., 2007, 180, 1595 CrossRef CAS.
- H. Huppertz and B. von der Eltz, J. Am. Chem. Soc., 2002, 124, 9376 CrossRef CAS.
- H. Huppertz, Z. Naturforsch., Teil B, 2003, 58, 278 CAS.
- H. Emme and H. Huppertz, Z. Anorg. Allg. Chem., 2002, 628, 2165 CrossRef.
- H. Emme and H. Huppertz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, i29 CrossRef.
- H. Emme and H. Huppertz, Chem.–Eur. J., 2003, 9, 3623 CrossRef CAS.
- H. Huppertz, S. Altmannshofer and G. Heymann, J. Solid State Chem., 2003, 170, 320 CrossRef CAS.
- H. Emme and H. Huppertz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, i23 CrossRef.
- J. S. Knyrim, F. Roeßner, S. Jakob, D. Johrendt, I. Kinski, R. Glaum and H. Huppertz, Angew. Chem., Int. Ed., 2007, 46, 9097 CrossRef CAS.
- S. C. Neumair, R. Glaum and H. Huppertz, Z. Naturforsch., Teil B, 2009, 64, 883 CAS.
- S. C. Neumair and H. Huppertz, Z. Naturforsch., Teil B, 2010, 65 Search PubMed , in press.
- Y. Wu, J.-Y. Yao, J.-X. Zhang, P.-Z. Fu and Y.-C. Wu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, i45 CrossRef.
- S. Jin, G. Cai, W. Wang, M. He, S. Wang and X. Chen, Angew. Chem., Int. Ed., 2010, 49, 4967 CrossRef CAS.
- A. C. Wright, Phys. Chem. Glasses, 2010, 51, 1.
- J. S. Knyrim, F. M. Schappacher, R. Pöttgen, J. Schmedt auf der Günne, D. Johrendt and H. Huppertz, Chem. Mater., 2007, 19, 254 CrossRef CAS.
- F. Pan, G. Shen, R. Wang, X. Wang and D. Shen, J. Cryst. Growth, 2002, 241, 108 CrossRef CAS.
- A. Perloff and S. Block, Acta Crystallogr., 1966, 20, 274 CrossRef CAS.
- D. L. Corker and A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 260 CrossRef.
- K.-I. Machida, G.-Y. Adachi and J. Shiokawa, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2008 CrossRef.
- J. S. Knyrim and H. Huppertz, J. Solid State Chem., 2007, 180, 742 CrossRef CAS.
- J. S. Knyrim and H. Huppertz, Z. Naturforsch., Teil B, 2008, 63, 707 CAS.
- F. Demartin, A. Minaglia and C. M. Gramaccioli, Can. Mineral., 2001, 39, 1105 CrossRef CAS.
- C. A. Gressler and J. E. Shelby, J. Appl. Phys., 1988, 64, 4450 CrossRef CAS.
- E. V. Grishchuk, N. P. Efryushina, V. P. Dotsenko and E. R. Gubanova, Inorg. Mater. (Transl. of Neorg. Mater.), 1998, 34, 520 Search PubMed.
- J. E. Shelby and L. D. Baker, Phys. Chem. Glasses, 1998, 39, 23 CAS.
- L. R. P. Kassab, L. C. Courrol, A. S. Morais, S. H. Tatumi, N. U. Wetter and L. Gomes, J. Opt. Soc. Am. B, 2002, 19, 2921 Search PubMed.
- C. K. C. K. Jayasankar, V. Venkatramu, P. Babu, T. Troster, W. Sievers, G. Wortmann and W. B. Holzapfel, J. Appl. Phys., 2005, 97, 093523 CrossRef.
- W. A. Pisarski, J. Pisarska, M. Maczka and W. Ryba-Romanowski, J. Mol. Struct., 2006, 792–793, 207 CrossRef CAS.
- A. V. Ravi Kumar, B. Apparao and N. Veeraiah, Bull. Mater. Sci., 1998, 21, 341 Search PubMed.
- G. Su and H. Toratani, Jpn. Kokai Tokkyo Koho, 1997, 6 Search PubMed.
- T. Suzuki, M. Hirano and H. Hosono, J. Appl. Phys., 2002, 91, 4149 CrossRef CAS.
- A. Haberer, R. Kaindl, J. Konzett, R. Glaum and H. Huppertz, Z. Anorg. Allg. Chem., 2010, 636, 1326 CrossRef CAS.
- A. Haberer, M. Enders, R. Kaindl and H. Huppertz, Z. Naturforsch., Teil B, 2010, 65 Search PubMed , in press.
- A. Haberer and H. Huppertz, J. Solid State Chem., 2009, 182, 888 CrossRef CAS.
- G. Corbel, R. Retoux and M. Leblanc, J. Solid State Chem., 1998, 139, 52 CrossRef CAS.
- E. Antic-Fidancev, G. Corbel, N. Mercier and M. Leblanc, J. Solid State Chem., 2000, 153, 270 CrossRef CAS.
- H. Müller-Bunz and T. Schleid, Z. Anorg. Allg. Chem., 2002, 628, 2750 CrossRef.
- A. Haberer, R. Kaindl and H. Huppertz, J. Solid State Chem., 2010, 183, 471 CrossRef CAS.
- A. Haberer, R. Kaindl, O. Oeckler and H. Huppertz, J. Solid State Chem., 2010, 183 Search PubMed , in press.
- B. G. Hyde, A. N. Bagshaw, S. Andersson and M. O'Keeffe, Annu. Rev. Mater. Sci., 1974, 4, 43 CrossRef CAS.
- D. J. M. Bevan, J. Mohyla, B. F. Hoskins and R. J. Steen, Eur. J. Solid State Inorg. Chem., 1990, 27, 451 CAS.
- J. P. Laval, A. Taoudi, A. Abaouz and B. Frit, J. Solid State Chem., 1995, 119, 125 CrossRef CAS.
- H. Müller-Bunz, O. Janka and T. Schleid, Z. Anorg. Allg. Chem., 2007, 633, 37 CrossRef.
- J. W. McCauley, P. Patel, M. Chen, G. Glide, E. Strassburger, B. Paliwal, K. T. Ramesh and D. P. Dandekar, J. Eur. Ceram. Soc., 2009, 29, 223 CrossRef CAS.
- P. Verdier and R. Marchand, Rev. Chim. Minér., 1976, 13, 145 CAS.
- J. Nishizawa and I. Shiota, US Pat., 4331737-14, 1982 Search PubMed.
- I. Shiota, N. Miyamoto and J. Nishizawa, Surf. Sci., 1979, 86, 272 CrossRef CAS.
- I. Shiota and J. Nishizawa, Surf. Interface Anal., 1979, 1, 185 CrossRef CAS.
- E. Soignard, D. Machon, P. F. McMillan, J. Dong, B. Xu and K. Leinenweber, Chem. Mater., 2005, 17, 5465 CrossRef CAS.
- I. Kinski, G. Miehe, G. Heymann, R. Theissmann, R. Riedel and H. Huppertz, Z. Naturforsch., Teil B, 2005, 60, 831 CAS.
- C. E. Zvoriste, S. A. Hering, I. Kinski, R. Riedel and H. Huppertz, Z. Naturforsch., Teil B, 2009, 64, 1115.
- H. Huppertz, S. A. Hering, C. E. Zvoriste, S. Lauterbach, O. Oeckler, R. Riedel and I. Kinski, Chem. Mater., 2009, 21, 2101 CrossRef CAS.
- J. W. McCauley, J. Am. Ceram. Soc., 1978, 61, 372 CAS.
- I. Adams, T. R. AuCoin and G. A. Wolff, J. Electrochem. Soc., 1962, 109, 1050 CrossRef CAS.
- A. M. Lejus, Rev. Hautes Temp. Réfract., 1964, 1, 53 Search PubMed.
- C. E. Zvoriste, L. Dubrovinsky, S. A. Hering, H. Huppertz, R. Riedel and I. Kinski, Int. J. High Pressure Res., 2009, 29, 389 Search PubMed.
Footnote |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| This journal is © The Royal Society of Chemistry 2011 |