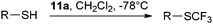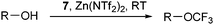DOI:
10.1039/C0CC02265A
(Feature Article)
Chem. Commun., 2011,
47, 102-115
Benziodoxole-based hypervalent iodine reagents for atom-transfer reactions†
Received
30th June 2010
, Accepted 4th August 2010
First published on 27th August 2010
Abstract
In the last decades, hypervalent iodine reagents have raised from chemical curiosities to mainstream reagents in organic synthesis. The use of benziodoxole-derived reagents has been especially successful in oxidation methods, whereas non-cyclic iodinanes have been used both for oxidation and atom-transfer reactions. On the other hand, the exceptional properties of benziodoxole reagents for atom-transfer reactions have only started to attract the attention of the synthetic community more recently. In this review, progress in the use of these compounds for C–X and C–C bond formations will be presented. In particular, recent breakthroughs in trifluoromethylation and alkynylation reactions have been realized since 2006 based on benziodoxole-derived reagents and these results are the main focus of this article.

Jonathan P. Brand
| Jonathan Brand was born in Strasbourg, France in 1984. He studied chemistry at the Ecole Nationale Supérieure de Chimie de Montpellier (France) and was graduated in 2008. He was awarded the “GlaxoSmithKline Certificate for Outstanding Academic Achievement in Organic Chemistry. 2007–2008 Academic Year”. His education included almost two years of internships at Actelion (Allschwil, Switzerland), GlaxoSmithKline (Harlow, UK) and Hoffmann-La Roche (Basel, Switzerland). Since 2008, he is carrying out his PhD at Ecole Polytechnique Fédérale de Lausanne (Switzerland) working on the direct alkynylation of heterocycles using hypervalent iodine reagents under the direction of Prof. Jérôme Waser. |

Davinia Fernández González
| Davinia Fernández González was born in Tui, Pontevedra, Spain in 1980. She studied at the Faculty of Chemistry at Santiago de Compostela where she obtained her diploma in Organic Chemistry in 2007. During her studies she was awarded an international fellowship that allowed her to spend 18 months in the EPF Lausanne where she worked in the synthesis and organic applications of new Ruthenium complexes. She is currently in the third year of her PhD studies in the Ecole Polytechnique Fédérale de Lausanne (Switzerland) working on the development of new alkynylation reactions under the direction of Prof. Jérôme Waser. |

Stefano Nicolai
| Stefano Nicolai was born in Civitavecchia (Italy) in 1979. He accomplished his studies in chemistry at the Faculty of Mathetical, Physical and Natural Sciences at the University of Rome 1 “La Sapienza” where he obtained his diploma in Organic Chemistry in 2007. In 2008, he worked at the same University on the synthesis of derivatives of the Wieland–Miescher ketones. He is currently in the second year of his PhD studies in the Ecole Polytechnique Fédérale de Lausanne (Switzerland) working on the application of hypervalent iodine chemistry to Wacker cyclization reactions under the direction of Prof. Jérôme Waser. |

Jérôme Waser
| Jérôme Waser was born in Sierre, Valais, Switzerland in 1977. He studied chemistry at ETH Zurich and obtained his Diploma in 2001. In 2002, he started his PhD studies at ETH Zurich with Prof. Erick M. Carreira, working on the development of metal-catalyzed amination reactions of olefins. In 2006, he joined Prof. Barry M. Trost at Stanford University and accomplished the total synthesis of Pseudolaric Acid B, a diterpene natural product. Since October 2007, he is working as tenure-track assistant professor at EPF Lausanne, focusing on the development and application of catalytic methods for the synthesis of bioactive compounds. |
1. Introduction
Hypervalent iodine compounds have always fascinated chemists, due to their non-classical bond character and exceptional reactivity.1 Nevertheless, the research in this area has been mostly curiosity-driven for several decades, and it is only more recently that general synthetic applications have emerged. For example, the Dess–Martin periodinane (DMP) as a mild and non-toxic reagent is now one of the most often used oxidant in organic synthesis.2 Structurally, it belongs to the benziodoxole-derived λ5iodine compounds, which are characterized by the inclusion of the iodine atom inside a ring.3 This cyclic structure confers to the benziodoxole-derived reagents an exceptional stability when compared to other hypervalent iodines, and their use for oxidation reactions is now well-established.4 A recent important progress in this field was the introduction of methods catalytic in iodine.5
Besides simple electron-transfer, the use of hypervalent iodine for oxidative atom transfer reactions is also emerging as a promising area, both for hetero- and carbon- atom transfer reactions.6 In contrast to oxidation reactions, non-cyclic reagents have been used in the vast majority of cases. In fact, one may argue that the lower reactivity of benziodoxole-based reagents is an obstacle for their use as oxidative functionalization reagents. In the case of highly unstable structures or for the development of catalytic methods, less reactive reagents are desirable, however. Despite promising preliminary results mostly for heteroatom transfer, the number of benziodoxole-based reagents used in functionalization reactions other than simple oxidations constitutes only a fraction of the reported structures and most of them are based on the λ3iodine benziodoxolone structure (Fig. 1). Whereas the formation of C–C bond using non-cyclic iodonium reagents has been very successful in the last decades,6c it is only in 2006 that Togni and co-workers reported the first use of benziodoxole-derived reagents 7 and 11 for CF3 transfer.7 In 2009, our group introduced ethynylbenziodoxolone (EBX) reagents 8 for acetylene transfer reactions.8 In these two recent works, key for success was the cyclic structure of the reagent, revealing for the first time the unique properties of benziodoxol(on)es for C–C bond formation.
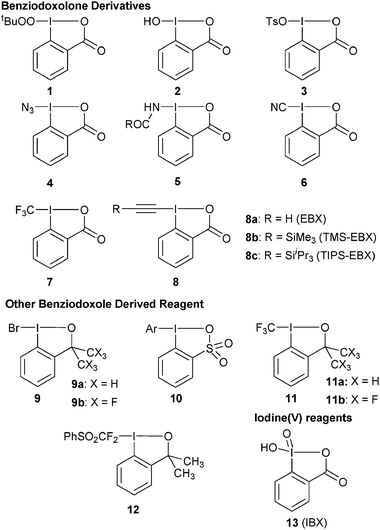 |
| | Fig. 1
Benziodoxole-based reagents used in atom-transfer reactions. | |
The purpose of this review is to summarize recent results in the use of benziodoxole-based reagents for atom-transfer processes. Reactions involving isolated benziodoxole-based reagents will be treated in priority and methods with in situ formation or “pseudo-benziodoxole” structures with only weak intramolecular interactions will not be covered in details. As a review on benziodoxole-derived reagents has appeared in 2005,3b focus will be on the results obtained in the last five years. We will begin with heteroatom transfer reactions (Chapter 2) and then continue with reactions involving C–C bond formation (Chapter 3), with a particular focus on CF3 (Section 3.1) and acetylene transfer (Section 3.2) reactions.
2. C–Heteroatom bond formation
A selection of early examples on the use of benziodoxole-derived reagents for heteroatom transfer reactions is presented in Scheme 1.3Tert-butylperoxy benziodoxolone 1 was introduced by Ochiai and co-workers (A).9 This reagent was efficient for the oxygenation of ethers, amines, sulfide and phenols. The peroxy group was conserved only in the case of phenols. The ammonium salt 14 of hydroxy benziodoxolone 2 has been used for the epoxidation of α,β-unsaturated ketones (B).10Tosyloxy benziodoxolone 3 has been applied for the iodotosylation of triple bonds in the presence of iodine (C).11 Interestingly, a chiral pseudo benziodoxolone has also been reported for the asymmetric bis-tosylation of double bonds and the α-tosyloxylation of ketones.12Nitrogen-transfer reactions were especially studied by Zhdankin and co-workers.13 Both the azidation (D) and amidation (E) of hydrocarbons was possible at high temperature via a radical pathway.
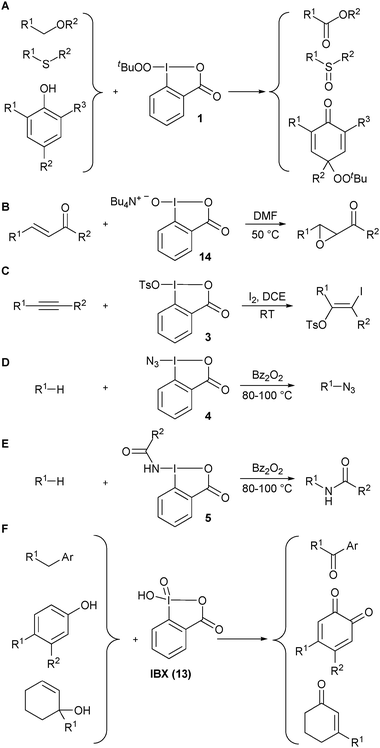 |
| | Scheme 1 Selected examples of heteroatom-transfer reactions before 2005. | |
λ5 iodanes have been mostly used for electron transfer. Nevertheless, IBX (13) has also emerged as an efficient reagent for the oxygenation of benzylic C–H bonds,14phenols15 and the oxidative rearrangement of allylic alcohols (F).16 Furthermore, several methods involving in situ formation of the active reagent have also been reported.3b
Since 2005, there are surprisingly few reports about the use of λ3 iodanes for heteroatom transfer reactions. Karade and co-workers studied the tosyloxylation of ketones with reagent 3 in details (eqn (1)).17 Good yields were obtained for aromatic and aliphatic ketones, as well as for ketoesters. Several other examples of tosyloxylation reactions using λ3 or λ5-benziodoxole-based compounds were reported, but in these cases the active reagent was formed in situ.18
| | 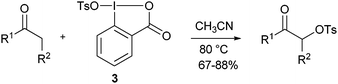 | (1) |
Based on earlier results of Martin and Amey,
19 Braddock and co-workers improved the synthesis of bromo benziodoxole
9a.
20 They demonstrated that
9a was an efficient source of bromonium ion for the bromination of
anisole (
15) and the bromolactonization of
4-pentenoic acid (
16) (
eqn (2)).
| | 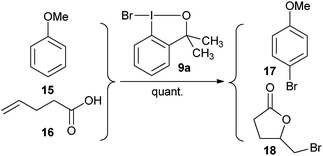 | (2) |
The use of IBX (
13) for oxygen-transfer and
oxidation of
phenol has been further investigated.
4b,21 In particular, Quideau and co-workers used SIBX (a stabilized formulation of IBX (
13))
22 in 2007 in a biomimetic synthesis of aquaticol (
20) (
Scheme 2).
21a
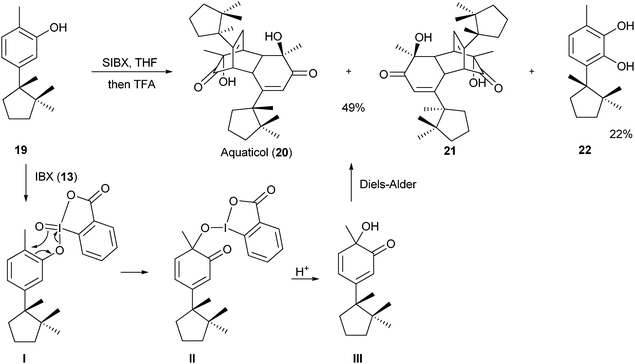 |
| | Scheme 2 Dearomatization-cycloaddition of phenols for the synthesis of aquaticol (20). | |
The treatment of (−)-hydroxycuparene (19) with SIBX led to oxidationvia intermediates I and II. Due to the presence of the methyl group, further oxidation to the quinone was not possible, and a very reactive diene III was obtained, which dimerized via a Diels–Alder cycloaddition. The desired natural product 20 was obtained in 49% yield as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with diastereoisomer 21. The main side product observed in the reaction was diol 22 resulting from the lack of regioselectivity in the oxidation step. A similar oxidative dimerization had been proposed for the biosynthesis of aquaticol (20).
:1 mixture with diastereoisomer 21. The main side product observed in the reaction was diol 22 resulting from the lack of regioselectivity in the oxidation step. A similar oxidative dimerization had been proposed for the biosynthesis of aquaticol (20).
In 2009, an important breakthrough was reported by Quideau and co-workers, who achieved asymmetric induction using a in situ generated chiral benziodoxolone reagent (eqn (3)).21e When the reaction was run with 2-methyl naphthol (23), the oxidation product 25 was more stable, and dimerization did not occur. Interestingly, epoxy alcohol 26 was obtained in excellent yield when the reaction was run with a catalytic amount of iodide 24 and an excess of m-CPBA (eqn (4)). Both a iodine (III) or an iodine (V) mechanisms were proposed for this reaction.21e In 2009, a similar approach based on an oxazolidinone pseudo-benziodoxole reagent has also been reported by Birman and co-workers.23 These exciting preliminary results set the bases for further catalytic asymmetric reactions based on chiral benziodoxole-derived reagents.
| | 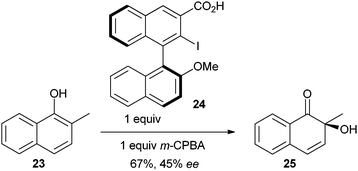 | (3) |
| |  | (4) |
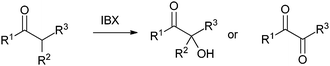
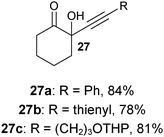
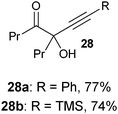
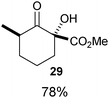
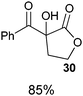
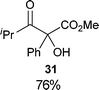
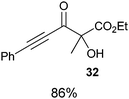
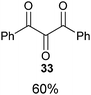
Several examples of the oxygenation of aliphatic C–H bonds using IBX (
13) have also appeared since 2005.
24 In particular, Kirsch and co-workers studied the hydroxylation of acidic C–H bonds α to carbonyl groups.
24a,b If a second activating group such as an
alkyne or an
ester was present,
hydroxylation became favored over the oxidation to the conjugated system usually observed with IBX (
13) (
Table 1). Methyne groups were oxidized to the corresponding
alcohols, whereas methylenes gave the fully oxidized carbonyls. Another example of α-hydroxylation of a
ketone using IBX (
13) was reported by Nagasawa and co-workers in the total synthesis of
saxitoxin (
eqn (5)).
24c In this case, the reaction was discovered serendipitously when attempting the oxidation of
alcohol 35.
Hydroxylation happened immediately after
oxidation of the
alcohol to the ketone.
| | 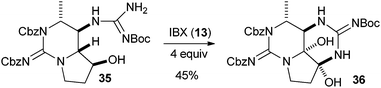 | (5) |
Other examples of the use of IBX for oxygen-transfer reactions include benzylic oxidation,
25oxidation of
oximes,
26 and an interesting oxidative rearrangement of homo-propargylic
alcohols to give
Z-enediones (
Scheme 3).
27 This reaction was proposed to occur
viaoxidation to the corresponding
ketone and α-hydroxylation, followed by a formal sigmatropic rearrangement to give the
Z-enedione after release of hydroxy benziodoxolone
2. The delivery of the
proton to the less hindered face of
allene intermediate
III led selectively to the
Z product.
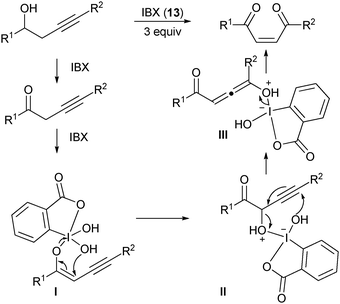 |
| | Scheme 3 Oxidative rearrangement of homopropargylic alcohols using IBX (13). | |
Table 1 Selected examples of the α-hydroxylation of carbonyls
3. C–C bond formation
Benziodoxole-based reagents have been much less investigated for C–C bond formation than non-cyclic hypervalent iodine salts. In particular, important progress in direct C–H functionalization reactions has been achieved using aryliodonium salts.6 A classical use of phenyl benziodoxolone 37 is the generation of benzyne under heating (eqn (6)).28| | 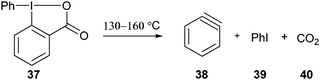 | (6) |
The use of hydroxy benziodoxolone 2 in Suzuki coupling has also been shortly examined (eqn (7)).29 Interestingly, no extra base was needed when using this reagent.| | 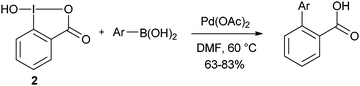 | (7) |
Preliminary results on the metal-mediated reactivity of the sulfur-analogous 10a of aryl benziodoxolones have been reported by Justik and co-workers (Scheme 4).30 The Cu-mediated reaction with oxygen or nitrogen nucleophiles led to the transfer of the sulfate-substituted aromatic ring, whereas the Pd-catalyzed reaction led to the formation of the biaryl product 42 incorporating the more electron-rich anisole ring. To the best of our knowledge, the reactivity of aryl benziodoxolones in classical cross-coupling reactions or direct C–H functionalization reactions has never been studied in details.
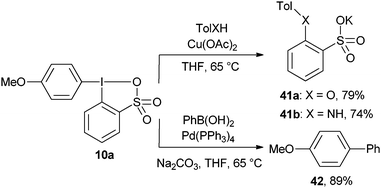 |
| | Scheme 4 Divergent reactivity of reagent 10a. | |
The synthesis and use of cyano benziodoxolone 6 has been reported by Zhdankin and co-workers (eqn (8)).31 A radical-like behaviour was observed at high temperature, allowing the cyanation of dimethyl aniline derivatives.
| | 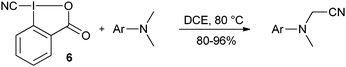 | (8) |
In fact, the rare and limited examples of
cyano or
aryl transfer using
benziodoxole-derived reagents stand in contrast with the impressive results obtained with non-cyclic iodonium salts.
6 However, this situation is completely different when we consider CF
3-transfer reactions or direct alkynylations of heterocycles: for both reactions, the use of
benziodoxole-derived reagents was essential for success. These two transformations will now be discussed in details.
3.1 CF2X-transfer
The introduction of fluorinated alkanes into organic compounds is an important field of research, due to the utility of these functional groups in medicinal chemistry.32 In particular, electrophilic sources of perfluoroalkyls are rare.33 Consequently, the discovery and development of electrophilic CF2X-benziodoxol(on)e reagents constituted an important progress in this area.
3.1.1
Trifluoromethylation
.
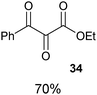
Togni and co-workers reported the first synthesis of trifluoromethyl benziodoxolone 7 and benziodoxole 11 in 2006 (Scheme 5).34 The corresponding non-cyclic reagents were not stable and could not be isolated. The synthesis of both reagents involved formal substitution with nucleophilic CF3 (generated in situ from Me3SiCF3) on activated benziodoxol(on)es 43 and 45. This first generation synthesis gave access to reagents 7 and 11 in respectively three and five steps from 2-iodobenzoic acid (46).
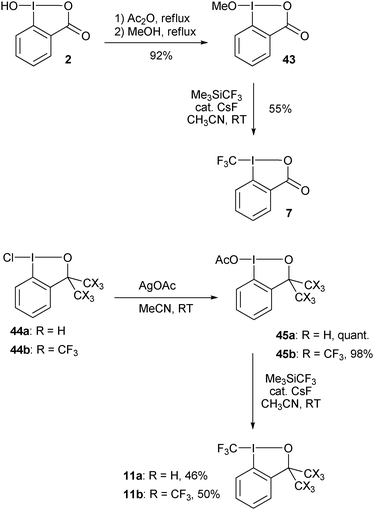 |
| | Scheme 5 First generation synthesis of 7 and 11. | |
More practical syntheses were reported later. Benziodoxolone 7 was accessed in 76% yield over three steps from 2-iodo benzoic acid (46) (eqn (9)).35 An improved one pot synthesis of 11a starting from benziodoxole 44a was also developed, affording the desired product 11a in 89% yield (eqn (10)).36
| | 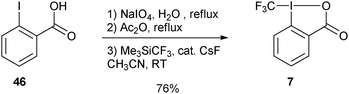 | (9) |
| | 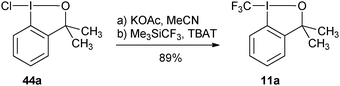 | (10) |
The use of reagent
11a was first examined for the introduction of CF
3 group onto cyclic β-ketoesters under phase transfer conditions (
eqn (11)).
34,36 The reaction is also efficient without phase transfer
catalyst, but more side reactions were observed. The reaction could be extended to α-nitro
esters using CuCl as
catalyst (
eqn (12)).
36 An important advantage of
benziodoxole 11a is the generation of the corresponding
alcohol during the reaction, which can be recycled for the synthesis of
11a.
| | 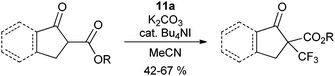 | (11) |
| | 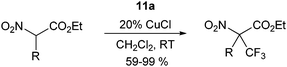 | (12) |
Trifluoromethylation of
thiols using reagent
11a lead to a convenient access to the SCF
3 functionality (
Table 2).
36 Both aromatic and aliphatic
thiols were used successfully. The reaction was carried out at −78 °C to avoid
disulfide formation. A high tolerance towards functional groups was observed, including highly functionalized substrates such as
thiosugars and
cysteine. This method was further extended to the selective trifluoromethylation of
cysteine in
peptides using
MeOH and
water as
solvent.
37
|
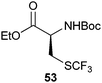
|
| 82% |
82% |
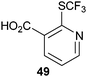
|
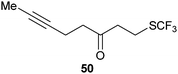
|
| 95% |
53% |
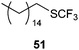
|
|
| 51% |
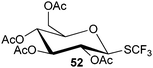
90% |
|
|
| 82% |
99% |
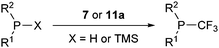
Trifluoromethylation of both secondary and primary phosphines was also investigated (Table 3).38 Both benziodoxolone 7 and benziodoxole 11a were efficient in this process, and 11a was more reactive. Aromatic and aliphatic phosphines could be used. Steric hindrance on the phosphine was detrimental for the reaction with both 7 and 11a. Interestingly, deprotonated phosphines (MPPh2, M = Li or K) only afforded traces of product. This observation led the authors to exclude phosphides as intermediates in the reaction. When Cy2PH was reacted with 7, the formation of CyP(CF3)2 and CyP(CF3)H in trace amounts was observed in addition to the major product Cy2PCF3. The attack of CF3-radical on Cy2PH was proposed to lead to C-P homolytic cleavage in addition to C–H cleavage. Based on these results, the authors proposed a radical pathway for the trifluoromethylation of phosphines.
| Substrate |
Conditions |
Product |
Yielda |
|
Isolated yield.
Conversion based on 19F NMR with PhCF3 as internal reference.
|
|
Ph2PH (55) |
7, RT |
Ph2P(CF3) (56) |
78 |
|
Ph2PH (55) |
11a, −78 °C to RT |
Ph2P(CF3) (56) |
74 |
|
Ph2PSiMe3 (57) |
7, RT |
Ph2P(CF3) (56) |
92b |
|
Ph2PSiMe3 (57) |
11a, −78 °C to RT |
Ph2P(CF3) (56) |
69 |
|
PhPH2 (58) |
7
, RT |
PhPH(CF3) (59) |
84b |
|
(o-Tol)2PH (60) |
7, RT |
(o-Tol)2P(CF3) (61) |
48 |
|
(o-Tol)2PH (60) |
11a, −78 °C to RT |
(o-Tol)2P(CF3) (61) |
50 |
Togni and co-workers applied the developed methodology to the synthesis of a P-bis(trifluoromethyl) derivative 62 (Fig. 2) of BINAP using reagent 11a.39 The addition of DBU was beneficial for the double trifluoromethylation. Unfortunately, ferrocene-derived ligands could not be functionalized, as they were too sensitive to oxidation.
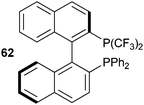 |
| | Fig. 2
Bis-trifluoromethyl-BINAP
62. | |
The trifluoromethylation of 2,4,6-trimethylphenol did not only afford the expected product 64, but also the dearomatized products 65 and 66 along with other minor side products (eqn (13)).35 Unfortunately, it was not possible to optimize the selectivity of the reaction. Two type of mechanisms have been proposed to explain this result. The phenolate formed from 63 could first form a charge-transfer complex with 11a, which then underwent single-electron transfer (SET). Radical recombination led then to the observed products. The second possible mechanism involved ligand exchange of benzoate with phenolate on I. CF3 could then be released and attack the phenolate ring. Unfortunately, mechanism investigation did not allow to distinguish between these pathways.
| | 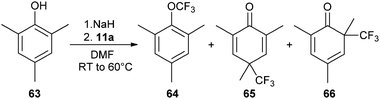 | (13) |
In order to extend the scope of trifluoromethylation reactions, Togni and co-workers examined the Lewis and Brønsted acid
activation of benziodoxol(on)es more in details. The development of the trifluoromethylation of
aliphatic alcohols catalyzed by zinc (II) salt resulted from these investigations (
Table 4).
40 The zinc salts were used in stoichiometric amount compared to
7 together with an excess of
alcohol. Catalytic amounts of zinc salts could be used, but they afforded lower yields. Togni proposed as a first step the ring opening of
7 with zinc salts to form complex
I which was detected by
ESI-MS (
Scheme 6). The authors then proposed either a nucleophilic attack of ROH on
I, followed by reductive elimination or a direct nucleophilic substitution to form complex
III. Complex
I is then regenerated by exchanging
2-iodobenzoic acid (
46) for
7. Interestingly, the authors showed that the reaction can also be catalyzed by a Brønsted acid (Tf
2NH) with lower efficiency.
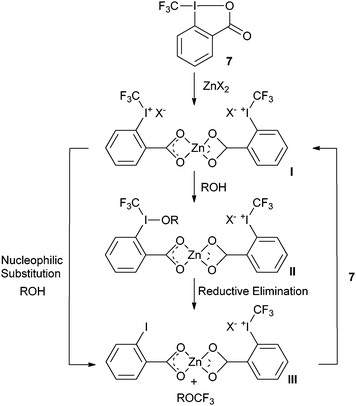 |
| | Scheme 6 Proposed mechanism for the trifluoromethylation of alcohols. | |
Table 4 Selected examples of trifluoromethylations of alcohols
The Brønsted acid activation of 7 was further used by Togni for the trifluoromethylation of sulfonic acids (eqn (14)).41 Strong acids were required and the use of the toluenesulfonate salts failed to give the product. Interestingly, the Hammett plot showed that the substituent on the sulfonic acid had only a minor effect on the reaction rate. In addition, the tetrahydrofuran ring was opened and trifluoromethylethers were formed when benziodoxolone 7 was used in presence of Lewis or Brønsted acids in THF.42
| | 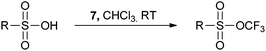 | (14) |
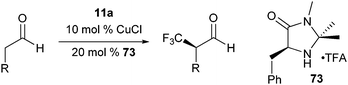
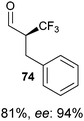
Mac Millan and co-workers recently reported the combination of organo- and Lewis acid- catalysis for the asymmetric α-trifluoromethylation of
aldehydes using hypervalent iodine reagent
11a (
Table 5).
43 The use of Lewis acid was crucial to obtain the product in useful yield. A range of Lewis acids were efficient in the reaction. CuCl gave the best yield and enantioselectivity. The mechanism proposed included a Lewis acid mediated opening of the
benziodoxole reagent
11a (
Scheme 7). The chiral
enamine I formed from the
aldehyde and
imidazolidinone catalyst 73 then attacked the iodine atom. Reductive elimination finally led to C–C bond formation.
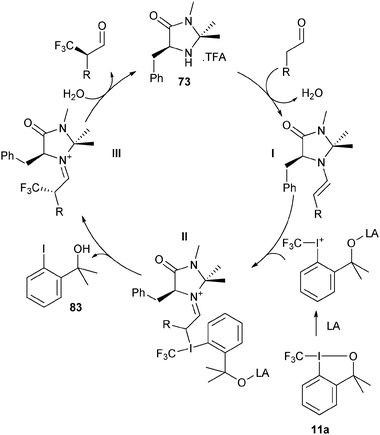 |
| | Scheme 7 Proposed mechanism for the organocatalytic trifluoromethylation of aldehydes. | |
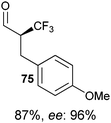
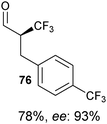
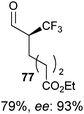
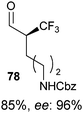
Table 5 Selected examples of enantioselective α-trifluoro-methylation of aldehydes
In contrast to arylation reactions,6 there are currently no successful examples of CF3-transfer using Pd or Cu catalysts and hypervalent iodine reagents.44
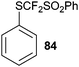
3.1.2. (Phenylsulfonyl)difluoromethylation.
Hu and coworkers reported the first synthesis of benziodoxole phenylsulfonyl difluoromethyl 12 using conditions analogous to Togni’s procedure (eqn (15)).45 The reaction was used for the (phenylsulfonyl) difluoromethylation of thiols (Table 6). High yields were obtained for both aryl and alkyl thiols. The obtained products were subjected to reductive desulfonylation to form the corresponding difluoromethylated sulfides.| | 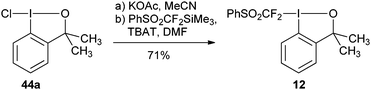 | (15) |
The chemistry of acetylenes has been intensively used in organic chemistry, material sciences and chemical biology.46 Classical methods for acetylene-transfer reactions usually involve the formation of acetylide anions. In contrast, the reverse approach using electrophilic alkynyl synthons has been much less developed.47 In this context, the use of alkynyl iodonium salts has been especially successful.47d,e Nevertheless, it had been limited to stoichiometric reactions with salts or organometallic reagents. We recently discovered that silyl-substituted ethynylbenziodoxolones (EBX (8)) are excellent acetylene transfer reagents, both in metal-free and metal-catalyzed reactions. These reagents are easily obtained in two steps from 2-iodo benzoic acid (46) on a 5–30 g scale (eqn (16)).48| | 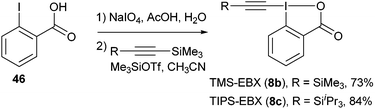 | (16) |
3.2.1 Alkynylation of acidic C–H bonds.
The alkynylation of tertiary acidic C–H bonds of α-substituted keto esters leads to the formation of all-carbon quaternary centers, which cannot be easily synthesized using classical acetylene chemistry. Prior to our work, this reaction was performed using alkynyliodonium salts in a two-steps procedure involving deprotonation with a strong base. Only a limited scope of substrates had been studied.49
Table 6 Selected examples of (phenylsulfonyl)-difluoromethylation of thiols
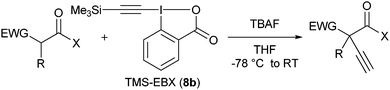
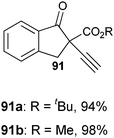
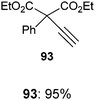
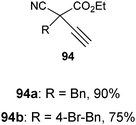
With the goal of developing an asymmetric alkynylation method, we initially examined phase transfer conditions developed by Jørgensen and co-workers50 with non-cyclic alkynyliodonium salts.51 However, low yields and no enantioselectivity were obtained in this case. In contrast, the use of TMS-EBX (8b) with KF as a base led to significant asymmetric induction (40% of ee) in the formation of the free acetylene product (eqn (17)).
| | 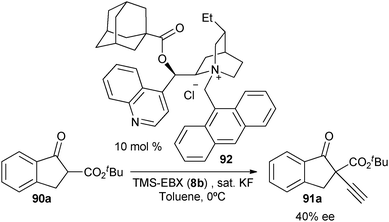 | (17) |
Unfortunately, further optimization of the enantioselectivity could not be achieved using this system, and we decided to investigate the racemic method more in details. Best yields were finally obtained using TBAF as a fluoride source in THF.
51 Under these conditions, efficient
acetylene transfer was observed, and full conversion could be obtained even at −78 °C (
Table 7).
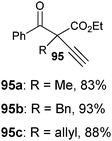
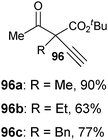
Table 7 Scope of the alkynylation of activated carbonyls
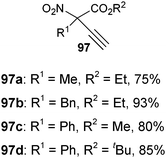
Cyclic keto esters and phenyl-diethylmalonate gave excellent yields in the alkynylation reaction. In contrast to cyclic substrates, linear keto-esters had never been investigated in the past. For these more challenging substrates, the desired acetylene products were obtained in 63–93% yield. Finally, the reaction could be extended to cyano and nitro esters, which are new classes of substrates for ethynylation reactions.
When the reaction was followed by 1H and 13C NMR, a fast deprotection of TMS-EBX (8b) was observed and EBX (8a) was obtained. EBX (8a) could be characterized at low temperature, but it decomposed when the temperature was risen over −20 °C. The use of 13C-labeled TIPS-EBX (8c) as reagent allowed us to observe a 1,2 shift of hydrogen in the product. This result indicated that a mechanism involving a conjugate addition to the alkyne, followed by α-elimination and 1,2- hydride shift via a carbene intermediate was probable. A similar mechanism had been proposed by Ochiai and co-workers in the case of alkynyliodonium salts.49a
3.2.2 C–H alkynylation of heterocycles.
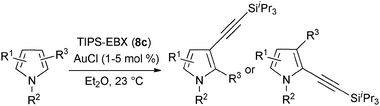
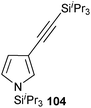
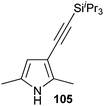
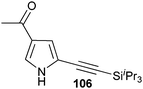
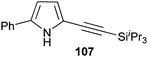
In contrast to the direct arylation and vinylation of heterocycles,52 direct alkynylation has been only scarcely investigated up to 2009.53 The field has attracted more interest since then and several direct alkynylations of heterocycles have been reported.47f,54 In 2009, we reported the use of TIPS-EBX (8c) in the gold-catalyzed direct alkynylation of indoles and pyrroles (Table 8).55 The reaction was run at room temperature under air and did not require dry solvents. Moreover, the 2-iodobenzoic acid (46) formed during the alkynylation reaction could be easily recovered via basic work-up and reused for the synthesis of TIPS-EBX (8c). The reaction was in general regioselective for the more electron-rich unsubstituted position of the heterocycles. The method was highly tolerant towards functionalities. Even highly reactive groups such as carboxylic acids, phenols or alcohols led to products in high yields. Importantly, the method was also orthogonal to classical cross-coupling, as halogen substituents were tolerated. The use of 2-substituted indoles was successful except for electron-withdrawing groups. In addition, pyrroles could also be alkynylated despite their high sensitivity. In case of pyrroles, the regioselectivity of the reaction could be inverted by the use of the bulky TIPS protecting group on the nitrogen to give 3-substituted pyrrole 104. Interestingly, the corresponding alkynyl iodonium salts did not afford any products, demonstrating the unique properties of the benziodoxolone structure.
|
86% |
|
76% |
| 67% |
| 93% |
| 85% |
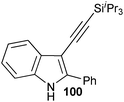
|
88% |
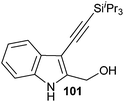
|
82% |
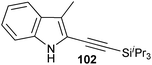
|
76% |
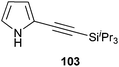
|
83% |
|
79% |
|
59% |
|
58% |
|
60% |
At least two mechanisms could be envisaged, in which Au acts either as a redox active metal or via π-activation (Scheme 8). The oxidative mechanism would include as a first step an oxidative addition of AuI on TIPS-EBX (8c) to form AuIII complex I. A ligand exchange with indole followed by reductive elimination would then give the alkynylation product. The π-activation mechanism would go through a gold catalyzed attack of indole on the triple bond of TIPS-EBX (8c). Depending on the regioselectivity of this process, either a β-elimination from IIIa or a α-elimination/1,2-shift from IIIb would lead to the product. A mechanism involving substitution and reductive elimination at iodine appeared less probable at this stage. Unfortunately, preliminary investigations did not allow to distinguish between these pathways.
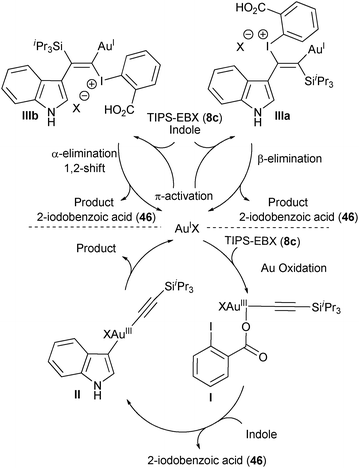 |
| | Scheme 8 Proposed mechanisms for the alkynylation of indole. | |
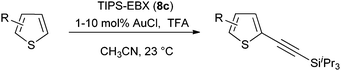
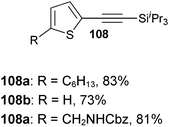
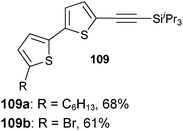
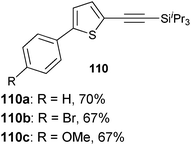
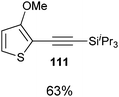
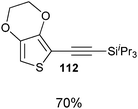
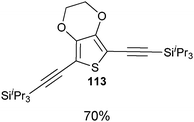
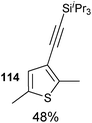
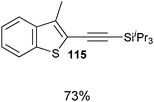
Both thiophenes and acetylenes are of utmost interest in organic materials. However, there are no methods for the direct alkynylation of thiophenes. The extension of the scope towards these heterocycles was consequently investigated. Unfortunately, previously developed conditions led to low yields and conversions, which is in accordance with the reported lower nucleophilicity of thiophenes. Nevertheless, we were delighted to discover that the addition of TFA (one equivalent compared to TIPS-EBX (8c)) gave good yields with a broad range of thiophenes (Table 9).56 This represented the first cooperative gold-Brønsted acid activation of alkynyl hypervalent iodine reagents. The reaction was tolerant towards a broad range of functional groups, except for electron-withdrawing groups directly attached to thiophene. Bithiophenes, phenylthiophenes and 3,4-ethylene-dioxythiophene, which are important in material sciences, could also be alkynylated. Finally, 3-methylbenzothiophene was also used successfully in the reaction.
Table 9 Selected examples of direct alkynylation of thiophenes
3.2.3 Intramolecular oxyalkynylation of olefins.
The encouraging results obtained with the alkynyl benziodoxolones motivated us to further explore their applicability in the field of Pd catalysis. In particular, the Wacker cyclization has been known as an effective method to generate O- and N-containing heterocycles starting from non-activated alkenes for nearly forty years.57 More recently, cascade reactions involving oxidative functionalizations of the key Pd-alkyl intermediate have been developed for C–C, C–X, C–O or C–N bond formation.58 Several of these processes have been proposed to proceed via a PdIV intermediate. The oxidative transfer of acetylene had never been realized using this approach, although Canty reported the use of alkynyliodonium salts to generate a PdIV-acetylene complex.59
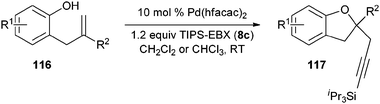
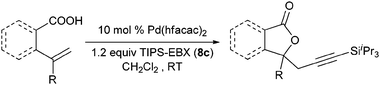
Our group recently described the first example of Pd-catalyzed intramolecular oxyalkynylation of non-activated olefins using TIPS-EBX (8c).60Pd hexafluoroacetyl acetonate was identified as the best catalyst for this process. TIPS-EBX (8c) was required for the reaction. Only very low yields were observed when using alkynyliodonum salts or less hindered silyl group on the acetylene.
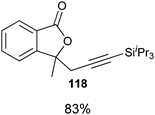
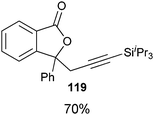
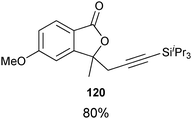
Good to very good yields were obtained with a broad range of o-allyl phenols (Table 10). Substrates bearing an electron-withdrawing group on the benzene ring afforded the highest yields. On the other hand, electron-donating groups were not tolerated. This could be due to the lower acidity of these substrates or their higher oxidation sensitivity.
Table 10 Scope of the alkynylation of o-allylphenols
| R1 = |
R2 = |
Yield |
| H (116a) |
Me (117a) |
73% |
|
4-Br (116b) |
Me (117b) |
68% |
|
4-CN (116c) |
Me (117c) |
76% |
|
4-NO2 (116d) |
Me (117d) |
72% |
|
4-MeCO (116e) |
Me (117e) |
87% |
|
3-Br (116f) |
Me (117f) |
66% |
|
2-Br (116g) |
Me (117g) |
52% |
| H (116h) |
H (117h) |
41% |
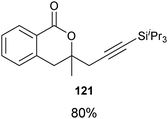
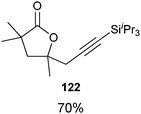
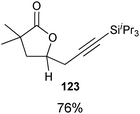
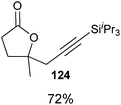
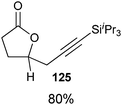
Indeed, it was found that carboxylic acids, which are more acidic and not oxidation sensitive, are good substrates for the reaction (Table 11). Aromatic γ- and δ-lactones could be obtained in good yields. Moreover, it was possible to extend our method to aliphatic carboxylic acids. To our delight, C–C bond formation was faster than β-hydride elimination in the case of 4-pentenoic acids and the alkynylation proceeded in good yield to give lactones 123 and 125.
The first steps of the catalytic cycle are probably olefin activation and nucleophilic addition to generate PdII alkyl σ-complex III (Scheme 9). These steps could proceed either via direct double bond activation or first via ligand exchange with the phenol to give Pd-alkoxide II. Complex III would then react with TIPS-EBX (8c), to afford PdIV complex IV. Reductive elimination would then occur with release of the product and formation of PdII complex V. In the last step, a ligand exchange is proposed to allow double bond activation.
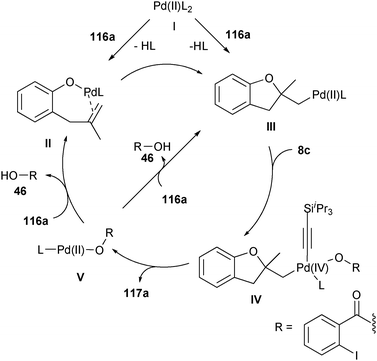 |
| | Scheme 9 Proposed mechanism for the oxyalkynylation of olefins. | |
4. Conclusions
In the field of hypervalent iodine chemistry, the use of benziodoxole-derived reagents has been mostly limited to oxidation reactions in the past. Functionalization reactions involving atom transfer from the reagent have been mostly limited to non-cyclic iodinanes. Despite promising results for hetero-atom transfer, it is only very recently that the unique properties of benziodoxole-derived reagents have been used for C–C bond formations, especially for the introduction of fluorinated alkanes and acetylenes. These promising preliminary results open new opportunities for the use of hypervalent iodine reagents in challenging oxidative functionalization reactions. The field is still in its infancy, and further work is required to develop new reactions based on benziodoxole-derived reagents. Particularly challenging will be the development of asymmetric methods or the use of processes catalytic in iodine with in situ formation of the active reagent.
5. Addendum (July 26, 2010)
During the reviewing process of this manuscript, a new overview article on electrophilic trifluoromethylation methods including hypervalent iodine reagents has appeared.61 Togni and co-workers have published a structural study of several new benziodoxole-based reagents.62 Furthermore, they have reported a detailed study on the trifluoromethylation of arenes.63 A broad scope of aromatic compounds was tested, including pyrroles, indoles, imidazoles, pyridines, phenols, anisoles and anilines. A positive effect of additives such as Zn salts or tris(trimethylsilyl)silyl chloride was observed. This work constitutes the broadest study of the electrophilic trifluoromethylation of arenes so far and demonstrated further the high potential of benziodoxole-based reagents. Nevertheless, the yields were highly dependent on the substrate structure, and further improvements will be needed to develop a truly general trifluoromethylation method for arenes.
Acknowledgements
EPFL and SNF (Swiss Science National Foundation, grant number 200021_119810) are acknowledged for financial support.
Notes and references
- For general reviews on hypervalent iodine, see:
(a) P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS;
(b)
A. Varvoglis, Hypervalent Iodine in Organic Synthesis, Academic Press, San Diego, CA, 1997 Search PubMed;
(c) A. Varvoglis, Tetrahedron, 1997, 53, 1179 CrossRef CAS;
(d) A. Kirschning, Eur. J. Org. Chem., 1998, 2267 CrossRef CAS;
(e) T. Wirth and U. H. Hirt, Synthesis, 1999, 1271 CrossRef CAS;
(f) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS;
(g)
T. Wirth, M. Ochiai, V. V. Zhdankin, G. F. Koser, H. Tohma and Y. Kita, Top. Curr. Chem.: Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis, Springer, Berlin, 2003, vol. 224 Search PubMed;
(h) T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656 CrossRef CAS;
(i) R. M. Moriarty, J. Org. Chem., 2005, 70, 2893 CrossRef CAS;
(j) U. Ladziata and V. V. Zhdankin, Arkivoc, 2006, 9, 26;
(k) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS;
(l) V. V. Zhdankin, Arkivoc, 2009, 1, 1.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- For reviews on benziodoxole-derived reagents, see:
(a) V. V. Zhdankin, Rev. Heteroatom Chem., 1997, 17, 133 Search PubMed;
(b) V. V. Zhdankin, Curr. Org. Synth., 2005, 2, 121 Search PubMed.
- For reviews on the use of hypervalent iodine for oxidations, see:
(a) H. Tohma and Y. Kita, Adv. Synth. Catal., 2004, 346, 111 CrossRef CAS;
(b) S. Quideau, L. Pouysegu, A. Ozanne and J. Gagnepain, Molecules, 2005, 10, 201 Search PubMed;
(c) M. Ochiai, Chem. Rec., 2007, 7, 12 CrossRef CAS;
(d) U. Ladziata and V. V. Zhdankin, Synlett, 2007, 527 CAS;
(e) M. A. Ciufolini, N. A. Braun, S. Canesi, M. Ousmer, J. Chang and D. Chai, Synthesis, 2007, 3759 CrossRef CAS;
(f) S. Quideau, L. Pouysegu and D. Deffieux, Synlett, 2008, 467 CrossRef CAS;
(g) M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086 RSC;
(h) T. Dohi, M. Ito, N. Yamaoka, K. Morimoto, H. Fujioka and Y. Kita, Tetrahedron, 2009, 65, 10797 CrossRef CAS;
(i) L. Pouysegu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235 CrossRef CAS.
-
(a) R. D. Richardson and T. Wirth, Angew. Chem., Int. Ed., 2006, 45, 4402 CrossRef CAS;
(b) M. Ochiai and K. Miyamoto, Eur. J. Org. Chem., 2008, 4229 CrossRef CAS;
(c) T. Dohi and Y. Kita, Chem. Commun., 2009, 2073 RSC.
-
(a) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924 CrossRef CAS;
(b) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS;
(c) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS.
- See section 3.1.
- See section 3.2.
-
(a) M. Ochiai, T. Ito, H. Takahashi, A. Nakanishi, M. Toyonari, T. Sueda, S. Goto and M. Shiro, J. Am. Chem. Soc., 1996, 118, 7716 CrossRef CAS;
(b) M. Ochiai, D. Kajishima and T. Sueda, Heterocycles, 1997, 46, 71 CrossRef CAS;
(c) M. Ochiai, A. Nakanishi and T. Ito, J. Org. Chem., 1997, 62, 4253 CrossRef CAS;
(d) M. Ochiai, A. Nakanishi and A. Yamada, Tetrahedron Lett., 1997, 38, 3927 CrossRef CAS;
(e) M. Ochiai, D. Kajishima and T. Sueda, Tetrahedron Lett., 1999, 40, 5541 CrossRef CAS;
(f) T. Sueda, S. Fukuda and M. Ochiai, Org. Lett., 2001, 3, 2387 CrossRef CAS.
- M. Ochiai, A. Nakanishi and T. Suefuji, Org. Lett., 2000, 2, 2923 CrossRef.
- T. Muraki, H. Togo and M. Yokoyama, J. Org. Chem., 1999, 64, 2883 CrossRef CAS.
- U. H. Hirt, M. F. H. Schuster, A. N. French, O. G. Wiest and T. Wirth, Eur. J. Org. Chem., 2001, 1569 CrossRef CAS.
-
(a) V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, M. S. Formaneck and J. T. Bolz, Tetrahedron Lett., 1994, 35, 9677 CrossRef CAS;
(b) V. V. Zhdankin, A. P. Krasutsky, C. J. Kuehl, A. J. Simonsen, J. K. Woodward, B. Mismash and J. T. Bolz, J. Am. Chem. Soc., 1996, 118, 5192 CrossRef CAS;
(c) V. V. Zhdankin, M. McSherry, B. Mismash, J. T. Bolz, J. K. Woodward, R. M. Arbit and S. Erickson, Tetrahedron Lett., 1997, 38, 21 CrossRef CAS.
- K. C. Nicolaou, P. S. Baran and Y. L. Zhong, J. Am. Chem. Soc., 2001, 123, 9313 CrossRef CAS.
- D. Magdziak, A. A. Rodriguez, R. W. Van De Water and T. R. R. Pettus, Org. Lett., 2002, 4, 285 CrossRef CAS.
- M. Shibuya, S. Ito, M. Takahashi and Y. Iwabuchi, Org. Lett., 2004, 6, 4303 CrossRef CAS.
- N. N. Karade, G. B. Tiwari, S. V. Shinde, S. V. Gampawar and J. M. Kondre, Tetrahedron Lett., 2008, 49, 3441 CrossRef CAS.
-
(a) R. D. Richardson, J. M. Zayed, S. Altermann, D. Smith and T. Wirth, Angew. Chem., Int. Ed., 2007, 46, 6529 CrossRef CAS;
(b) U. S. Mahajan and K. G. Akamanchi, Synlett, 2008, 983 CrossRef;
(c) A. Shah, Z. A. Khan, N. Choudhary, C. Loholter, S. Schafer, G. P. L. Marie, U. Farooq, B. Witulski and T. Wirth, Org. Lett., 2009, 11, 3578 CrossRef CAS;
(d) U. Farooq, S. Schafer, A. A. Shah, D. M. Freudendahl and T. Wirth, Synthesis, 2010, 1023 CAS.
- R. L. Amey and J. C. Martin, J. Org. Chem., 1979, 44, 1779 CrossRef CAS.
- D. C. Braddock, G. Cansell, S. A. Hermitage and A. J. P. White, Chem. Commun., 2006, 1442 RSC.
-
(a) J. Gagnepain, F. Castet and S. Quideau, Angew. Chem., Int. Ed., 2007, 46, 1533 CrossRef CAS;
(b) J. Gagnepain, R. Mereau, D. Dejugnac, J. M. Leger, F. Castet, D. Deffieux, L. Pouysegu and S. Quideau, Tetrahedron, 2007, 63, 6493 CrossRef CAS;
(c) N. Lebrasseur, J. Gagnepain, A. Ozanne-Beaudenon, J. M. Leger and S. Quideau, J. Org. Chem., 2007, 72, 6280 CrossRef CAS;
(d) L. Pouysegu, M. Marguerit, J. Gagnepain, G. Lyvinec, A. J. Eatherton and S. Quideau, Org. Lett., 2008, 10, 5211 CrossRef CAS;
(e) S. Quideau, G. Lyvinec, M. Marguerit, K. Bathany, A. Ozanne-Beaudenon, T. Buffeteau, D. Cavagnat and A. Chenede, Angew. Chem., Int. Ed., 2009, 48, 4605 CrossRef CAS;
(f) A. H. Wu, Y. Z. Duan, D. W. Xu, T. M. Penning and R. G. Harvey, Tetrahedron, 2010, 66, 2111 CrossRef CAS;
(g) R. Bernini, E. Mincione, F. Crisante, M. Barontini and G. Fabrizi, Tetrahedron Lett., 2009, 50, 1307 CrossRef CAS;
(h) R. Bernini, F. Crisante, M. Barontini and G. Fabrizi, Synthesis, 2009, 3838 CrossRef CAS;
(i) R. Bernini, M. Barontini, F. Crisante, M. C. Ginnasi and R. Saladino, Tetrahedron Lett., 2009, 50, 6519 CrossRef CAS.
- A. Ozanne, L. Pouysegu, D. Depernet, B. Francois and S. Quideau, Org. Lett., 2003, 5, 2903 CrossRef CAS.
- J. K. Boppisetti and V. B. Birman, Org. Lett., 2009, 11, 1221 CrossRef CAS.
-
(a) S. F. Kirsch, J. Org. Chem., 2005, 70, 10210 CrossRef CAS;
(b) A. Duschek and S. E. Kirsch, Chem.–Eur. J., 2009, 15, 10713 CrossRef CAS;
(c) O. Iwamoto, H. Koshino, D. Hashizume and K. Nagasawa, Angew. Chem., Int. Ed., 2007, 46, 8625 CrossRef CAS.
- L. R. Ojha, S. Kudugunti, P. P. Maddukuri, A. Kommareddy, M. R. Gunna, P. Dokuparthi, H. B. Gottam, K. K. Botha, D. R. Parapati and T. K. Vinod, Synlett, 2009, 117 CAS.
- A. Souto, J. Rodriguez and C. Jimenez, Tetrahedron Lett., 2009, 50, 7395 CrossRef CAS.
- B. Crone and S. F. Kirsch, Chem. Commun., 2006, 764 RSC.
-
(a) F. M. Beringer and I. Lillien, J. Am. Chem. Soc., 1960, 82, 725 CrossRef CAS;
(b) F. M. Beringer and S. J. Huang, J. Org. Chem., 1964, 29, 445 CAS.
- M. Xia and Z. C. Chen, Synth. Commun., 2000, 30, 63 CrossRef CAS.
- M. W. Justik, J. D. Protasiewicz and J. B. Updegraff, Tetrahedron Lett., 2009, 50, 6072 CrossRef CAS.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz, B. Mismash, J. K. Woodward and A. J. Simonsen, Tetrahedron Lett., 1995, 36, 7975 CrossRef CAS.
-
(a)
K. Uneyama, Organofluorine Chemistry, Blackwell Publishing, Oxford, 2006 Search PubMed;
(b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef.
- T. Umemoto, Chem. Rev., 1996, 96, 1757 CrossRef CAS.
- P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579 CrossRef CAS.
- K. Stanek, R. Koller and A. Togni, J. Org. Chem., 2008, 73, 7678 CrossRef CAS.
- I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754 CrossRef CAS.
- S. Capone, I. Kieltsch, O. Flogel, G. Lelais, A. Togni and D. Seebach, Helv. Chim. Acta, 2008, 91, 2035 CrossRef CAS.
- P. Eisenberger, I. Kieltsch, N. Armanino and A. Togni, Chem. Commun., 2008, 1575 RSC.
- N. Armanino, R. Koller and A. Togni, Organometallics, 2010, 29, 1771 CrossRef CAS.
- R. Koller, K. Stanek, D. Stolz, R. Aardoom, K. Niedermann and A. Togni, Angew. Chem., Int. Ed., 2009, 48, 4332 CrossRef CAS.
- R. Koller, Q. Huchet, P. Battaglia, J. M. Welch and A. Togni, Chem. Commun., 2009, 5993 RSC.
- S. Fantasia, J. M. Welch and A. Togni, J. Org. Chem., 2010, 75, 1779 CrossRef CAS.
- A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 4986 CrossRef CAS.
- Recently, Yu and co-workers developed a Pd-catalyzed direct trifluoromethylation of arenes, but sulfur-based reagents were superior to hypervalent iodine: X. Wang, L. Truesdale and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 3648 Search PubMed.
- W. Zhang, J. M. Zhu and J. B. Hu, Tetrahedron Lett., 2008, 49, 5006 CrossRef.
-
F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material Science, Wiley-VCH, Weinheim, 2005 Search PubMed.
- For a few selected references, see:
(a) S. I. Miller and J. I. Dickstein, Acc. Chem. Res., 1976, 9, 358 CrossRef CAS;
(b) A. S. Kende, P. Fludzinski, J. H. Hill, W. Swenson and J. Clardy, J. Am. Chem. Soc., 1984, 106, 3551 CrossRef CAS;
(c) M. G. Moloney, J. T. Pinhey and E. G. Roche, J. Chem. Soc., Perkin Trans. 1, 1989, 333 RSC;
(d) T. Kitamura, K. Nagata and H. Taniguchi, Tetrahedron Lett., 1995, 36, 1081 CrossRef CAS;
(e) V. V. Zhdankin and P. J. Stang, Tetrahedron, 1998, 54, 10927 CrossRef CAS;
(f) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096 CrossRef CAS , and references therein.
-
TIPS-EBX (8c) will soon be commercially available from Sigma-Aldrich (catalog number 728365) V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz and A. J. Simonsen, J. Org. Chem., 1996, 61, 6547 Search PubMed.
-
(a) M. Ochiai, T. Ito, Y. Takaoka, Y. Masaki, M. Kunishima, S. Tani and Y. Nagao, J. Chem. Soc., Chem. Commun., 1990, 118 RSC;
(b) M. D. Bachi, N. Barner, P. J. Stang and B. L. Williamson, J. Org. Chem., 1993, 58, 7923 CrossRef CAS.
- T. B. Poulsen, L. Bernardi, J. Aleman, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2007, 129, 441 CrossRef CAS.
- D. Fernández González, J. P. Brand and J. Waser, Chem.–Eur. J., 2010, 16, 9457 CrossRef.
-
(a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC;
(b) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS;
(c) F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269 CrossRef CAS.
-
(a) K. Kobayashi, M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 8528 CrossRef CAS;
(b) R. Amemiya, A. Fujii and M. Yamaguchi, Tetrahedron Lett., 2004, 45, 4333 CrossRef CAS;
(c) B. A. Trofimov, Z. V. Stepanova, L. N. Sobenina, A. I. Mikhaleva and I. A. Ushakov, Tetrahedron Lett., 2004, 45, 6513 CrossRef CAS;
(d) I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742 CrossRef CAS.
- Selected examples:
(a) M. Tobisu, Y. Ano and N. Chatani, Org. Lett., 2009, 11, 3250 CrossRef CAS;
(b) N. Matsuyama, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 4156 CrossRef CAS;
(c) F. Besselievre and S. Piguel, Angew. Chem., Int. Ed., 2009, 48, 9553 CrossRef CAS;
(d) Y. H. Gu and X. M. Wang, Tetrahedron Lett., 2009, 50, 763 CrossRef CAS;
(e) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef CAS.
- J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS.
- J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2010, 49 DOI:10.1002/anie.201003179.
- For recent reviews, see:
(a)
J. Tsuji, Palladium Reagents and Catalysts, John Wiley and Sons, Chichester, 2004, p. 27 Search PubMed;
(b)
P. M. Henry, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, John Wiley and Sons, New York, 2002, p. 2119 Search PubMed;
(c)
T. Hosokawa and S.-I. Murahashi, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, John Wiley and Sons, New York, 2002, p. 2141 Search PubMed;
(d) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS;
(e) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS;
(f) B. M. Stoltz, Chem. Lett., 2004, 33, 362 CrossRef CAS;
(g) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS.
- For a few selected examples, see:
(a) E. J. Alexanian, C. Lee and E. J. Sorensen, J. Am. Chem. Soc., 2005, 127, 7690 CrossRef CAS;
(b) L. V. Desai and M. S Sanford, Angew. Chem., Int. Ed., 2007, 46, 5737 CrossRef CAS;
(c) Y. Li, D. Song and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2962 CrossRef CAS.
-
(a) A. J. Canty, J. Patel, T. Rodemann, J. H. Ryan, B. W. Skelton and A. H. White, Organometallics, 2004, 23, 3466 CrossRef CAS;
(b) A. J. Canty, T. Rodemann, B. W. Skelton and A. H. White, Organometallics, 2006, 25, 3996 CrossRef CAS;
(c) A. J. Canty, M. G. Gardiner, R. C. Jones, T. Rodemann and M. Sharma, J. Am. Chem. Soc., 2009, 131, 7236 CrossRef CAS.
- S. Nicolai, S. Erard, D. Fernández González and J. Waser, Org. Lett., 2010, 12, 384 CrossRef CAS.
- N. Shibata, A. Matsnev and D. Cahard, Beilstein J. Org. Chem., 2010, 6 DOI:10.3762/bjoc.6.65.
- K. Niedermann, J. M. Welch, R. Koller, J. Cvengros, N. Santschi, P. Battaglia and A. Togni, Tetrahedron, 2010, 66, 5753 CrossRef CAS . The issue 31 of Tetrahedron 2010 (pages 5727–5918) was dedicated to hypervalent iodine chemistry.
- M. S. Wiehn, E. V. Vinogradova and A. Togni, J. Fluorine Chem., 2010, 131, 951 CrossRef CAS.
Footnote |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.