Facile synthesis of planar chiral N-oxides and their use in Lewis base catalysis†‡§
J. Robin
Fulton
a,
Jean E.
Glover
b,
Lamin
Kamara
a and
Gareth J.
Rowlands
*ab
aDepartment of Chemistry, University of Sussex, Falmer, BN1 9QJ, UK
bInstitute of Fundamental Sciences – Chemistry, Massey University, Private Bag 11 222, Palmerston North, New Zealand. E-mail: g.j.rowlands@massey.ac.nz; Fax: +64 6 350 5682; Tel: +64 6 356 9099 extn 3566
First published on 24th September 2010
Abstract
A rapid and versatile method for the preparation of planar chiral [2.2]paracyclophane-derived pyridines and pyridine N-oxides is reported. The potential utility of these compounds in Lewis base catalysis is briefly introduced.
Chiral heteroaromatics are a valuable motif in enantioselective catalysis and related disciplines.1–4 Planar chiral heterocycles are uncommon, with the majority based on ferrocene or related complexes. Chiral cyclophane-based heteroaromatics is a field ripe for exploration.4–8 We are interested in the synthesis of planar chiral [2.2]paracyclophane derivatives9 and in this communication, we disclose a rapid route to planar chiral pyridine N-oxides and reveal their potential as Lewis base catalysts.
A multitude of [2.2]paracyclophane-derived heterocycles have been reported, but have found little utility as the syntheses are either convoluted or of limited generality.5 To overcome this limitation, we sought a versatile route to rapidly prepare 2-([2.2]paracyclophan-4-yl)pyridines 1 (Scheme 1). Pyridines and pyridine N-oxides have an excellent pedigree as ligands for catalysis, photoelectronic dyes and metal–organic frameworks as well as being common organocatalyst.1,2,10,11 2-([2.2]Paracyclophan-4-yl)pyridines and related compounds are known; Hopf has reported a simple route to a pyridinyl[2.2]paracyclophane by a nitrone cycloaddition, but the generality of this reaction has not been assessed,12 whilst Pfaltz et al.6 and Andrus et al.8 have accessed derivatives of [2](1,4)benzeno[2](2,5)pyridinophane in generally low yields.
![Possible syntheses of 2-([2.2]paracyclophane-4-yl)pyridines.](/image/article/2011/CC/c0cc02216k/c0cc02216k-s1.gif) | ||
| Scheme 1 Possible syntheses of 2-([2.2]paracyclophane-4-yl)pyridines. | ||
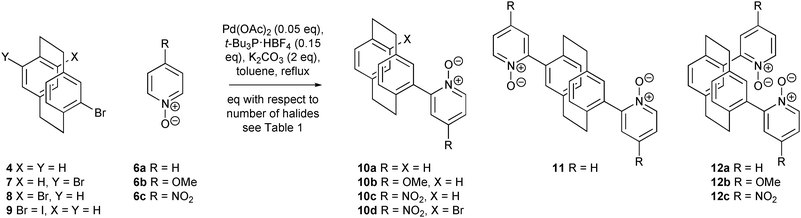
The simplest route to 2-([2.2]paracyclophan-4-yl)pyridines 1 involves the cross coupling of suitably functionalised [2.2]paracyclophane derivative 2 or 4 with its complementary 2-pyridinyl unit 3 or 5 (Scheme 1); frustratingly, such chemistry is problematic. Whilst 2-halopyridines 3 readily participate in the cross coupling reactions, the corresponding 4-[2.2]paracyclophanyl boronic acids 2 are unstable.13 Reversing the functionality fails to overcome this problem; 4-halo[2.2]paracyclophanes 4 are suitable precursors but 2-pyridyl boronic acids 5 are inherently unstable and rarely employed in cross-coupling reactions. The direct arylation chemistry of Fagnou et al. appeared to offer an effective solution to these limitations, permitting the coupling of 4-halo[2.2]paracyclophanes 4 and pyridine N-oxides 6.14–16
To evaluate the utility of this methodology in [2.2]paracyclophane chemistry, racemic 4-bromo[2.2]paracyclophane 4 and pyridine N-oxide 6a were treated with tri-tert-butylphosphonium tetrafluoroborate, palladium(II) acetate and potassium carbonate at reflux in toluene (Scheme 2; Table 1; Entry 1).§ The desired 10a was isolated in 66% yield with the remainder of the material comprising of [2.2]paracyclophane, the product of proto-debromination. A perfunctory attempt was made to optimise the reaction but the only improvement was obtained by increasing the catalyst loading. Use of the more reactive 4-iodo[2.2]paracyclophane with the addition of Ag2CO3 led to a slight improvement (Entry 4) but this did not offset the additional steps required to form the iodide from the bromide. Reducing the amount of pyridine N-oxide to less than 3 eq. results in a dramatic decrease in yield. The reaction appears general, with both the electron rich 4-methoxypyridine N-oxide 6b and the electron deficient 4-nitropyridine N-oxide 6c undergoing coupling in respectable yields (Entries 5 and 6). The initial attempt to form a bis(pyridine N-oxide) utilised pseudo-para4,16-dibromo[2.2]paracyclophane 7, thus minimising any inter-deck interactions. As expected a mixture of the desired bis(pyridine N-oxide) 11 and debromo mono(pyridine N-oxide) 10a was formed (Entry 7). More rewarding was the successful coupling of the pseudo-ortho4,12-dibromo[2.2]paracyclophane derivative 12. Under our standard reaction conditions, all three pyridine N-oxides, 6a, 6b and 6c, reacted to give mixtures of mono- and bis(pyridine N-oxides) (Entries 8, 9 and 10). Generally, the combined yields are excellent. It is unclear if the variation in ratio of mono- to bis(pyridine N-oxide) is due to steric hindrance or an electronic effect affecting the rate of proto-debromination. Interestingly, only in the reaction of more reactive nitro derivative16 was the bromo pyridine N-oxide observed (Entry 10). The optimum yield of the bis(pyridine N-oxide) 11/12 was obtained by doubling the number of equivalents of all reagents. Currently, the major limitation to this methodology is associated with purification; all the products and by-products display limited solubility precluding simple chromatography or recrystallisation techniques. Purification withstanding, this chemistry is extremely practicable; it requires no special precautions, such as inert atmosphere, and permits the coupling of aryl bromides and pyridine N-oxides simply by heating in the presence of palladium(II) acetate and an air-stable phosphine salt.
 | ||
| Scheme 2 Synthesis of mono- and bis(pyridine N-oxides). See Table 1 for yields. | ||
| Entry | Halide | R | Product(s) (yield %) | |
|---|---|---|---|---|
| a Reaction employed Cy3P·HBF4 as ligand. b Reaction employed 8 eq. 6a, 0.1 eq. Pd(OAc)2, 0.3 eq. t-Bu3P·HBF4 and 8.0 eq. K2CO3. c 3.0 eq. 6a, 0.05 eq. Pd(OAc)2, 0.15 eq. t-Bu3P·HBF4, 0.5 eq. Ag2CO3, 2.0 eq. K2CO3. | ||||
| 1 | 4 | H | 66 (10a) | |
| 2a | 4 | H | 41 (10a) | |
| 3b | 4 | H | 74 (10a) | |
| 4c | 9 | H | 65 (10a) | |
| 5 | 4 | OMe | 62 (10b) | |
| 6 | 4 | NO2 | 59 (10c) | |
| 7 | 7 | H | 38 (10a) | 38 (11) |
| 8 | (R)-8 | H | 29 ((R)-10a) | 54 ((R)-12a) |
| 9 | (R)-8 | OMe | 53 ((R)-10b) | 32 ((R)-12b) |
| 10 | (±)-8 | NO2 | 32 ((±)-10c), 15 ((±)-10d) | 25 ((±)-12c) |
The mono- and bis(pyridine N-oxides) are readily modified to form various pyridines. Simple reduction of 10a and 12a is best achieved by treatment with trichlorosilane and triethylamine, although other methods also furnish the products (Scheme 3).15,16 The dipyridine 13 can be selectively oxidised to the mixed pyridine/N-oxide–pyridine 15 by reaction with mCPBA. The N-oxide functionality permits functionalisation of C6 of the pyridine moiety. Treatment of either 10a or 12a with diethylcarbamoyl chloride and trimethylsilyl cyanide17 results in the formation of the nitriles 17 and 14 in moderate yield. Simple acid hydrolysis furnishes potentially valuable chiral pyridine-2-carboxylic acids such as 18 (Scheme 3). Curiously, addition of the nitrile moiety to 11 has proven impossible under these conditions.
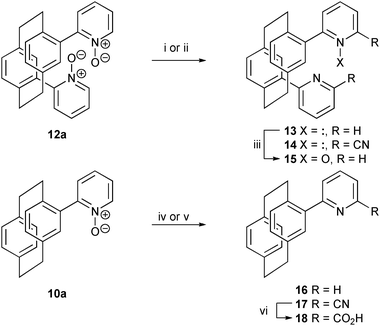 | ||
| Scheme 3 Reagents and conditions: (i) Cl3SiH (15 eq.), Et3N (10 eq.), CH2Cl2, reflux (13, 88%); (ii) TMSCN (2 × 3 eq.), diethylcarbamoyl chloride (2 × 3 eq.), CH2Cl2, rt (14, 51%); (iii) mCPBA (1.1 eq.), CH2Cl2, rt (15, 65%); (iv) Cl3SiH (15 eq.), Et3N (10 eq.), CH2Cl2, reflux (16, 80%); (v) TMSCN (2 × 3 eq.), diethylcarbamoyl chloride (2 × 3 eq.), CH2Cl2, rt (17, 60%); (vi) 6 M HCl, reflux (18, 93%). | ||
The utility of these novel N-oxides was demonstrated in the Lewis base-mediated allylation of benzaldehyde with allyltrichlorosilane.1,11,18,19 Enantiomerically pure mono- and bis(pyridine N-oxides) (R)-10a,b, (R)-12a,b and (R)-15 were prepared from (R)-4,12-dibromo[2.2]paracyclophane. For these preliminary tests, benzaldehyde 19 was treated with allyltrichlorosilane 20, diisopropylethylamine and 10 mol% N-oxide in CH2Cl2 at −78 °C (Scheme 4); no attempt was made to optimise the conditions. All the N-oxides tested displayed high activity and whilst only modest enantioselectivities were observed, the results exhibit an intriguing electronic effect. Both the mono- and bis-unsubstituted pyridine N-oxides (R)-10a and (R)-12a gave homoallylic alcohol (R)-21 with identical enantioselectivities (38% ee). The electron rich, methoxy substituted pyridine N-oxides (R)-10b and (R)-12b furnished the opposite enantiomer, (S)-21, with comparable enantioselectivity (36% ee and 28% ee, respectively). Most interestingly, the unsubstituted mixed pyridine/pyridine N-oxide 15 gave the opposite result compared to unsubstituted 12a providing (S)-21 with 30% ee. Simple steric factors do not explain these differences and we surmise that electronic factors play a crucial role.19 It is unlikely that either of the bis(pyridine N-oxides) are bidentate as they give the same enantioselectivity as their monodentate counterparts.
 | ||
| Scheme 4 Reagents and conditions: (i) N-oxide (R)-10a, 10b, 12a, 12b or 15 (0.1 eq.), iPr2NEt, CH2Cl2, −78 °C. | ||
In conclusion, we have outlined a new strategy for the preparation of planar chiral pyridines and pyridine N-oxides. The route is based on Fagnou's direct arylation methodology and permits the synthesis of these potentially valuable compounds in just two steps from [2.2]paracyclophane. These compounds can be considered our first generation of paracyclophane-based planar chiral Lewis base catalysts. Further studies will be directed at delineating the electronic effects in the allylation reaction and modifying the basic framework to form better Lewis base catalysts. The use of these compounds in other applications, such as palladacycle formation, will also be investigated and disclosed in due course.
We appreciated funding from Massey University (GJR), Massey University for a University Technicians Award (JEG), the EPSRC (EP/D50175X/1; LK) and the University of Sussex. We thank Chirotech Technology Ltd and KISCO Ltd for the donation of materials.
Notes and references
- G. Chelucci, G. Murineddu and G. A. Pinna, Tetrahedron: Asymmetry, 2004, 15, 1373 CrossRef CAS.
- A. V. Malkov and P. Kočovský, Curr. Org. Chem., 2003, 7, 1737 CrossRef CAS.
- V. Cesar, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534 CrossRef CAS; M. C. Perry and K. Burgess, Tetrahedron: Asymmetry, 2003, 14, 951 CrossRef CAS; A. Pfaltz, J. Heterocycl. Chem., 1999, 36, 1437 CAS; D. R. Snead, H. Seo and S. Hong, Curr. Org. Chem., 2008, 12, 1370 CrossRef CAS.
- G. C. Fu, Acc. Chem. Res., 2000, 33, 412 CrossRef CAS; G. C. Fu, Acc. Chem. Res., 2006, 39, 853 CrossRef CAS.
- A. A. Aly and A. B. Brown, Tetrahedron, 2009, 65, 8055 CrossRef CAS.
- U. Wörsdörfer, F. Vögtle, M. Nieger, M. Waletzke, S. Grimme, F. Glorius and A. Pfaltz, Synthesis, 1999, 597 CrossRef.
- N. Kanomata, J. Suzuki, H. Kubota, K. Nishimura and T. Enomoto, Tetrahedron Lett., 2009, 50, 2740 CrossRef CAS; W. Z. Duan, Y. D. Ma, H. Q. Xia, X. Y. Liu, Q. S. Ma and J. S. Sun, J. Org. Chem., 2008, 73, 4330 CrossRef CAS; Y. Matsuoka, Y. Ishida, D. Sasaki and K. Saigo, Chem.–Eur. J., 2008, 14, 9215 CrossRef CAS; G. Ricci and R. Ruzziconi, Tetrahedron: Asymmetry, 2005, 16, 1817 CrossRef CAS; J. G. Seitzberg, C. Dissing, I. Sotofte, P. O. Norrby and M. Johannsen, J. Org. Chem., 2005, 70, 8332 CrossRef; B. Tao, M. M. C. Lo and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 353 CrossRef CAS; U. Wörsdörfer, F. Vogtle, F. Glorius and A. Pfaltz, J. Prakt. Chem. (Weinheim, Ger.), 1999, 341, 445 Search PubMed.
- Q. Chai, C. Song, Z. J. Sun, Y. D. Ma, C. Q. Ma, Y. Dai and M. B. Andrus, Tetrahedron Lett., 2006, 47, 8611 CrossRef CAS.
- P. B. Hitchcock, A. C. C. Hodgson and G. J. Rowlands, Synlett, 2006, 2625 CAS; G. J. Rowlands and R. J. Seacome, Beilstein J. Org. Chem., 2009, 5 Search PubMed; G. J. Rowlands, Org. Biomol. Chem., 2008, 6, 1527 RSC; P. B. Hitchcock, G. J. Rowlands and R. J. Seacome, Org. Biomol. Chem., 2005, 3, 3873 RSC; P. B. Hitchcock, G. J. Rowlands and R. Parmar, Chem. Commun., 2005, 4219 RSC.
- R. P. Wurz, Chem. Rev., 2007, 107, 5570 CrossRef CAS; H. L. Kwong, H. L. Yeung, C. T. Yeung, W. S. Lee, C. S. Lee and W. L. Wong, Coord. Chem. Rev., 2007, 251, 2188 CrossRef CAS; R. Murugan and E. F. V. Scriven, Aldrichimica Acta, 2003, 36, 21 CAS.
- A. Malkov and P. Kočovský, Eur. J. Org. Chem., 2007, 29 CrossRef CAS; A. V. Malkov, M.-M. Westwater, A. Gutnov, P. Ramírez-López, F. Friscourt, A. Kadlčíková, J. Hodačová, Z. Rankovic, M. Kotora and P. Kočovský, Tetrahedron, 2008, 64, 11335 CrossRef CAS.
- H. Hopf, A. A. Aly, V. N. Swaminathan, L. Ernst, I. Dix and P. G. Jones, Eur. J. Org. Chem., 2005, 68 CrossRef CAS.
- A. J. Roche and B. Canturk, Org. Biomol. Chem., 2005, 3, 515 RSC; R. J. Seacome, D. Phil., University of Sussex, 2008.
- D. J. Schipper, L. C. Campeau and K. Fagnou, Tetrahedron, 2009, 65, 3155 CrossRef CAS; L. C. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35; L. C. Campeau and K. Fagnou, Chem. Soc. Rev., 2007, 36, 1058 RSC.
- L. C. Campeau, D. R. Stuart, J. P. Leclerc, M. Bertrand-Laperle, E. Villemure, H. Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291 CrossRef.
- L. C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020 CrossRef CAS.
- M. G. N. Russell, R. W. Carling, J. R. Atack, F. A. Bromidge, S. M. Cook, P. Hunt, C. Isted, M. Lucas, R. M. McKernan, A. Mitchinson, K. W. Moore, R. Narquizian, A. J. Macaulay, D. Thomas, S.-A. Thompson, K. A. Wafford and J. L. Castro, J. Med. Chem., 2005, 48, 1367 CrossRef CAS.
- P. Kwiatkowski, P. Mucha, G. Mlostoń and J. Jurczak, Synlett, 2009, 1757 CAS; A. Kadlčíková, R. Hrdina, I. Valterová and M. Kotora, Adv. Synth. Catal., 2009, 351, 1279 CrossRef CAS; D. E. Bergbreiter and D. Ortiz-Acosta, Tetrahedron Lett., 2008, 49, 5608 CrossRef CAS; J. Gawronski, N. Wascinska and J. Gajewy, Chem. Rev., 2008, 108, 5227 CrossRef CAS.
- A. V. Malkov, P. Ramírez-López, L. Biedermannová, L. Rulíšek, L. Dufková, M. Kotora, F. J. Zhu and P. Kočovský, J. Am. Chem. Soc., 2008, 130, 5341 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Keith Fagnou. |
| ‡ This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| § Electronic supplementary information (ESI) available: Full experimental for all new compounds along with copies of the 1H and 13C NMR spectra. See DOI: 10.1039/c0cc02216k |
| This journal is © The Royal Society of Chemistry 2011 |