Reversible morphological switching of nanostructures in solution†‡
Adam O.
Moughton§
,
Joseph P.
Patterson
and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: R.K.O-Reilly@warwick.ac.uk; Fax: +44 (0)24 7652 4112
First published on 31st August 2010
Abstract
The design and synthesis of a tuneable and reversible morphology switching copolymer system is reported. The kinetics of the transition under a range of conditions has been explored, as has the stabilization of the resultant structures.
Amphiphilic block copolymers (BCPs) are able to self-assemble into a range of 3-dimensional morphologies in aqueous solution, whose characteristics and size can be readily tuned by the BCP's chemical composition and physical properties.1–3 Well-defined and functional BCPs have been utilized as building blocks for the synthesis of nanosized architectures for use in a diverse range of new applications in nanotechnology.4–6 The ability for self-assembled structures to respond to changes in external stimuli such as pH,7–9 temperature10–12 or light13,14 is particularly important for their use in ‘smart’ applications such as stimulus triggered delivery vehicles, sensors or nanoreactors.5,11,15 Tailoring the design of BCPs for use in smart self-assembled architectures has been aided by advances in controlled radical polymerization (CRP) techniques. Of the various CRP techniques, reversible addition fragmentation chain transfer (RAFT) polymerization is perhaps the most versatile for the creation of well-defined and functional amphiphilic BCPs.16–18 Incorporating functionality onto the (α and/or ω) chain ends of the BCP is particularly facile via the R group of the RAFT chain transfer agent (CTA) or via post transformation of the Z group.19
In recent years, several research groups have reported the synthesis of BCP micelles which undergo a thermally induced morphology transition to form hollow vesicles.20–22 Aside from changing their 3D shape, a morphological transition also changes the nanostructure's sequestration properties, thus perhaps inducing encapsulation and/or release behavior of hydrophilic (and hydrophobic) substrates. Often BCPs which display a thermally induced morphology transition contain a thermo-responsive poly(N-isopropyl acrylamide, PNIPAM) block located between a low Tg hydrophobic block and a hydrophilic block. The lower critical solution temperature (LCST) transition of the PNIPAM block has been utilized to modify the amphiphilic balance of the copolymers which gives rise to a change in the packing parameter p leading to a change in the amphiphile's preferred solution morphology.1 Thermoswitching BCP systems studied to date often require extended times for transitions to occur and this has, to date, limited their further application. Thus new BCP systems with improved switching kinetics are of great current interest.
In the design of a new BCP structure we hoped to address the problems associated with previously reported systems to enable a faster and fully reversible transition. Towards this goal, the synthesis of a copolymer which is more readily soluble in water at room temperature along with a polymer with a low glass transition temperature was targeted compared to our previous BCP design.20 The BCP system studied also employs a chain end functional charged ‘head group’, to act as a permanently hydrophilic group to stabilize the structures when the BCP becomes completely hydrophobic above the LCST of the copolymer. Thus the diblock copolymer, 3, was synthesized using sequential RAFT polymerization techniques employing a ‘head-group’ functionalized RAFT CTA, 1 (see ESI‡). This gave rise to a well-defined chain end functionalized diblock copolymer, PMA27-b-PNIPAM47, 3 (MnNMR = 8.2 kDa, MnGPC,DMF = 9.3 kDa, Mw/Mn = 1.25) (Fig. 1). We determined the LCST of 3 to be 36 °C in D2O utilizing VT 1H NMR spectroscopy as previously described.23
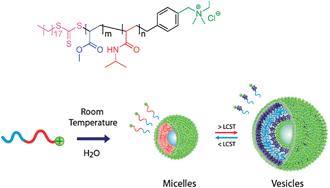 | ||
| Fig. 1 Recyclable thermally induced micelle–vesicle switching for a charged amphiphilic diblock copolymer, 3. | ||
The self-assembly of the diblock copolymer, 3, was first achieved via direct dissolution into a 1∶9 tetrahydrofuran (THF)/water mixture at a concentration of 1 mg mL−1. Dynamic light scattering (DLS) of the resultant aggregates, 4, at 25 °C showed the presence of rather ill-defined aggregates with a hydrodynamic diameter (Dh,app) of 31 nm (PDI = 0.34). Analysis of a dried sample of 4 at 25 °C, viatransmission electron microscopy (TEM), stained with uranyl acetate, confirmed the presence of spherical micelles, Davg = 20 ± 5 nm (Fig. 2). We also prepared micelles in 100% watervia dissolution of 3 into a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶9 THF/water mixture followed by exhaustive dialysis into deionised water. Dialysis gave a final solution of aggregates, 5, with a copolymer concentration of 0.71 mg mL−1 in pure water. These aggregates were proposed to be spherical micelles and had a Dh,app = 19 nm by number (PDI = 0.47) by DLS. The solution of 5 was analyzed by TEMvia drop deposition of a diluted sample at 25 °C and negative staining with uranyl acetate. The TEMs showed the presence of monodisperse spherical micelles with an average diameter, Davg = 17 ± 2 nm (Fig. 2). Zeta potential measurements were made on the micelles, 5 (ζ = +25 ± 8 mV at pH = 7) confirming that the micelles have a positive surface charge which is consistent with the ‘head-group’ being located at the outer micelle surface. Finally, 3 was self-assembled into 100% watervia direct dissolution to give micelles, 6, whose average diameter by DLS was 18 nm (PDI = 0.36), which is very close to the size obtained via the solvent switch method. For all nanostructures a small proportion of larger aggregates were observed in the intensity distribution from DLS.
∶9 THF/water mixture followed by exhaustive dialysis into deionised water. Dialysis gave a final solution of aggregates, 5, with a copolymer concentration of 0.71 mg mL−1 in pure water. These aggregates were proposed to be spherical micelles and had a Dh,app = 19 nm by number (PDI = 0.47) by DLS. The solution of 5 was analyzed by TEMvia drop deposition of a diluted sample at 25 °C and negative staining with uranyl acetate. The TEMs showed the presence of monodisperse spherical micelles with an average diameter, Davg = 17 ± 2 nm (Fig. 2). Zeta potential measurements were made on the micelles, 5 (ζ = +25 ± 8 mV at pH = 7) confirming that the micelles have a positive surface charge which is consistent with the ‘head-group’ being located at the outer micelle surface. Finally, 3 was self-assembled into 100% watervia direct dissolution to give micelles, 6, whose average diameter by DLS was 18 nm (PDI = 0.36), which is very close to the size obtained via the solvent switch method. For all nanostructures a small proportion of larger aggregates were observed in the intensity distribution from DLS.
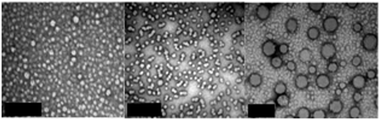 | ||
| Fig. 2 TEMs of micelles, 4 (LHS), 5 (middle) and 7 (RHS), drop deposited and stained with uranyl acetate (scale bar = 200 nm). | ||
It was proposed that the newly synthesized PMA27-b-PNIPAM47 diblock 3 would display similar thermomorphic behavior to that previously reported for a related diblock,20 when stirred and heated above its LCST. Furthermore, it was postulated that the structures would undergo a faster and more readily reversible transition due to the low Tg of the permanently hydrophobic block and the increased PNIPAM block length. To explore the thermomorphic behavior, initially a solution of micelles 4 in 1∶9 THF/water was subjected to heating and stirring at 65 °C with the evolution of the aggregate size being monitored by DLS analysis (Fig. 3). A dramatic change in the size of the aggregates (to ca. 140 nm) was observed after heating the solution for 23 h, thus suggesting a possible morphology change. Fig. 3 indicates that no significant change in micelle size was observed for the first 15 h of heating (perhaps due to vitrification of the PNIPAM block given its high Tg) after which time a steady increase in size over 10 h from ca. 40–140 nm was observed.
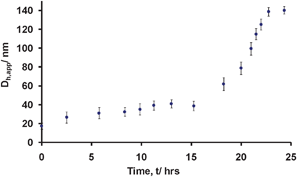 | ||
| Fig. 3 Apparent hydrodynamic diameter, Dh,app, of 4 upon stirring at 65 °C as a function of time. Error bars indicate the sample dispersity. | ||
The heated aggregates, 7, were proposed to be hollow vesicles, however, to elucidate their structure, the solution was analyzed viaTEM (Fig. 2). Upon deposition of the sample (at 65 °C) and staining with uranyl acetate, large aggregates with Davg = 85 ± 16 nm were observed. The diameters of the vesicles by TEM were lower than in solution by DLS. This is consistent with similar reports studying polymeric vesiclesviaTEM in the dried state.24 In addition, static light scattering techniques were used to determine the radius of gyration and this value in combination with the hydrodynamic radius (after 4 d at 65 °C) was used to confirm the hollow nature of 7, giving Rg/Rh = 0.99.25 Further confirmation of the size and distribution of 4 and 7 was obtained by AFM (see ESI‡).
However, upon TEM analysis, a population of smaller spherical aggregates could also be observed in the presence of the larger structures. We hypothesize that these are micelles due to the rapid reverse transition of the vesicles upon cooling during sample preparation.26 This infers that the reverse transition is much faster than the micelle–vesicle conversion. To confirm this the reversibility of the transition was investigated viaDLS and TEM measurements. After being heated for 27 h, 7 (Dh,app = 147 nm, PDI = 0.27) changed back into smaller aggregates, 4′ (Dh,app = 29 nm, PDI = 0.49) after cooling for 1 h at 25 °C. These aggregates are very similar in size and dispersity to the original micelles, 4, indicating a completely reverse and fast transition was occurring. To further highlight the robust nature of the morphology switching the size of the aggregates in solution was monitored by DLS over three successive morphology transition cycles (Fig. 4).
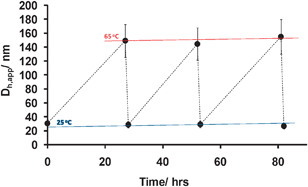 | ||
| Fig. 4 Apparent hydrodynamic diameters, Dh,app over time, upon heating a solution of 4 at 65 °C and cooling to 25 °C to afford 7 and repeating for 3 cycles. Error bars indicate the sample dispersity. | ||
Exploring the morphology transition in 100% water would be of particular interest, since it may be more applicable for drug and/or gene delivery uptake and release applications than mixed solvent systems. Thus, a solution of micelles, 5, in 100% water was subjected to heating and stirring at 65 °C as described for 4. The evolution in the aggregate size was monitored by DLS and the observations were similar to that reported for 4 in THF/water. This aqueous micelle solution also demonstrated reversibility and cycling. Given that the LCST of 3 was 36 °C, we proposed that the transition may be possible at lower temperatures. To test this, a solution of micelles, 5, in 100% water was stirred and heated at 40 °C with the evolution of the aggregate size followed viaDLS (see ESI‡). The transition at this temperature takes approximately five times longer as that at 65 °C and the vesicles, 9, were also larger in size, Dh,app = 218 nm and PDI = 0.41 after 114 h. The reason for these differences are not clear but could be due to slower transition kinetics or due to incomplete hydration of the PNIPAM block at this lower temperature. It is proposed that a lower temperature transition would be particularly attractive for drug or gene delivery applications. It should be noted that, the DLS studies, over the entire time course, were extremely reproducible, even over different runs, different concentrations and polymer batches.
One potential limitation of this system is the lack of control to ‘lock in’ a particular morphology to enable further control over the switching behavior. To explore this we investigated the effect of acid additives on the transition given the literature precedent for their interaction with PNIPAM.27 The effect of two biologically relevant additives on the transition at 40 °C was studied, L-ascorbic acid and citric acid. Upon repeating the switching experiment for 5, the addition of acid appeared to inhibit the transition, with no increase in nanostructure size or dispersity observed over 160 h (see ESI‡). This was not the expected result as it was proposed that the additive would lower the LCST, however, when one considers the mechanism of action of these additives in the dehydration of PNIPAM then this result may not be too surprising.28 Furthermore, since the acids have multiple H-bonding sites, interactions can occur intermolecularly as well as intramolecularly and will not only dehydrate the PNIPAM but will effectively crosslink the chains. We also confirmed by VT-NMR that the additives were not increasing the LCST of the polymer and thus preventing switching at 40 °C.
To further investigate the interaction of the additives within the nanostructures, zeta potential measurements were made on the ‘stabilized’ micelles. In the presence of 5 mg mL−1 of citric acid, the micelles gave zeta values of 36 ± 6 mV and with L-ascorbic acid, ζ = +35 ± 5 mV. These values are similar to that found for the initial micelles and hence support the theory that the additives do not bind to the charged surface of the micelle but that they reside within the PNIPAM corona. Further confirmation that the L-ascorbic acid was undergoing H-bonding with PNIPAM was obtained viaIR spectroscopy (see ESI‡). We also explored the effect of additives on micelles which had already transformed into vesicles, 7. Upon addition of L-ascorbic acid and cooling to room temperature a slight increase in nanostructure size was observed. After 3 days large structures (10) were still observed by DLS. This strongly suggests that the morphologies produced upon transition can be stabilized to lock in their structure. This provides a further level of nanostructure control which may be important in the realization of the potential applications of these materials.
In conclusion, we have shown that a charged block copolymer can undergo a relatively fast and fully reversible transition between micelle to vesicle morphologies. Furthermore, this new switching system is also very reproducible with the transition time consistent over 3 cycles. We have expanded the utility of this transition, showing that it can be performed at 40 °C and in mixed organic/aqueous or purely aqueous environments. This allows for the fine tuning of the transition time and could be useful in the design of drug or gene delivery applications. Furthermore, we have made initial investigations into the H-bonding stabilization of the micelles and vesicle structures using chemical additives. Such stabilized micelles or vesicles could be particularly useful for applications in biomedicine or materials science.
The EPSRC, Royal Society and University of Warwick are thanked for funding. Some of the equipment used in this work was obtained through Birmingham Science City with support from Advantage West Midlands and the ERDF.
Notes and references
- Y.-Y. Won, A. K. Brannan, H. T. Davis and F. S. Bates, J. Phys. Chem. B, 2002, 106, 3354–3364 CrossRef CAS.
- A. M. Nystrom, J. W. Barlets, W. Du and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1023–1037 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- D. M. Vriezema, M. Comellas Aragones, J. W. Elemans, J. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS.
- W. Meier, Chem. Soc. Rev., 2000, 29, 295–303 RSC.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- S. Liu and S. P. Armes, Angew. Chem., Int. Ed., 2002, 41, 1413–1416 CrossRef CAS.
- J. Rodriguez-Hernandez and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef CAS.
- Y. Li, B. S. Lokitz and C. L. McCormick, Macromolecules, 2006, 39, 81–89 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- P. Bhargava, Y. Tu, J. X. Zheng, H. Xiong, R. P. Quirk and S. Z. D. Cheng, J. Am. Chem. Soc., 2007, 129, 1113–1121 CrossRef CAS.
- F. D. Jochum, L. zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS.
- R. M. Uda, T. Tanabe, Y. Nakahara and K. Kimura, Soft Matter, 2008, 4, 560–563 RSC.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- A. O. Moughton and R. K. O'Reilly, Chem. Commun., 2010, 46, 1091–1093 RSC.
- A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130, 12264–12265 CrossRef CAS.
- H. Wei, C.-Y. Yu, C. Chang, C.-Y. Quan, S.-B. Mo, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, Chem. Commun., 2008, 4598–4600 RSC.
- J. Deng, Y. Shi, W. Jiang, Y. Peng, L. Lu and Y. Cai, Macromolecules, 2008, 41, 3007–3014 CrossRef CAS.
- A. Blanazs, M. Massignani, G. Battaglia, S. P. Armes and A. J. Ryan, Adv. Funct. Mater., 2009, 19, 2906–2914 CrossRef CAS.
- W. Burchard, Adv. Polym. Sci., 1983, 48, 1–124 CAS.
- C. Ott, R. Hoogenboom, S. Hoeppener, D. Wouters, J. F. Gohy and U. S. Schubert, Soft Matter, 2009, 5, 84–91 RSC.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS.
- L. B. Sagle, Y. Zhang, V. A. Litosh, X. Chen, Y. Cho and P. S. Cremer, J. Am. Chem. Soc., 2009, 131, 9304–9310 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for Chem Comm. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and further characterization for the polymers and assembled structures is provided. See DOI: 10.1039/c0cc02160a |
| § Current address: Department of Chemistry, University of Minnesota, 207 Pleasant St., SE Minneapolis, USA. |
| This journal is © The Royal Society of Chemistry 2011 |