Lyotropic liquid crystalline phases from helical β-peptides as alignment media†‡
Christina M.
Thiele
*a,
William C.
Pomerantz§
b,
Nicholas L.
Abbott
c and
Samuel H.
Gellman
b
aClemens Schöpf Institut für Organische Chemie und Biochemie, Technische Universität Darmstadt, Petersenstr. 22, 64287 Darmstadt, Germany. E-mail: cthiele@thielelab.de
bDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA
cDepartment of Chemistry, University of Wisconsin-Madison, Madison, WI 53706, USA
First published on 25th October 2010
Abstract
Lyotropic liquid crystalline (LLC) phases from helical β-peptides are proposed as novel water-based alignment media. In contrast to α-peptides, β-peptides form LLCs at very short chain lengths and at concentrations as low as 1 percent. Spectra obtained in these LLC phases are artefact-free and lead to RDCs of the desired size. First indications towards enantiodiscrimination are provided.
The determination of three dimensional structure by NMR spectroscopy usually relies on dihedral angles and distances from 3J couplings and NOEs, respectively.1 It has recently been shown that residual dipolar couplings (RDCs) can yield information complementary to these conventional NMR parameters for the investigation of structure and dynamics of biomolecules2 as well as for the determination of conformation and configuration of organic compounds.3
RDCs belong to the class of anisotropic NMR parameters. The molecule in question thus needs to be oriented with respect to the magnetic field. This is achieved with so called alignment or orienting media, two main classes of which exist: (lyotropic) liquid crystalline (LLC) phases2–4 and stretched polymer gels.2,3,5 To obtain RDCs of suitable size (tens of Hz) with high accuracy and precision it is necessary to be able to adjust the degree of order induced. For stretched polymer gels this can be done using a stretching device,6 while for LLC phases a variation in the concentration of the phase is usually applied.7 This approach, however, is possible only above the critical concentration (ccrit) of the LC phase. LLC phases based on α-peptides, with α-helical poly-γ-benzyl-L-glutamate (PBLG) and the enantiomer (PGDB) being the most prominent members, require rather high molecular weights (MW) to form stable LC phases and are usually characterized by quite high critical concentrations in organic solvents (>6 wt%).8 Thus, the degree of order induced tends to be very large, which complicates the extraction of RDCs measured in LLCs based on α-peptides.8
β-Peptides, such as 1,9,10 form stable LLC phases at very short chain lengths and at much lower ccrit relative to α-peptides (ccrit = 1 wt% for 1). The underlying design principles for 1 include use of the 14-helix-stabilizing cyclically constrained trans-2-aminocyclohexanecarboxylic acid (ACHC) residue and capping of the N-terminus with a heptanoyl moiety to promote self-assembly.9
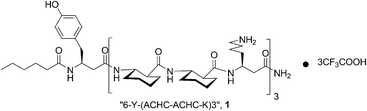
We wondered whether the low critical concentration of the LLC phases from such a helical β-peptide translates into a low induced degree of order for the solute, and whether the helical β-peptide would allow the observation of enantiodiscrimination as is the case with the much longer helical α-peptides.11 Thus we prepared a LLC phase comprised of β-peptide 1 (2.1% w/w in D2O), confirmed the formation of the LLC phase and its homogeneity by 2H NMR spectroscopy and added L-proline as solute (ΔνQ = 19.3 Hz for final sample).¶ We chose proline as a solute because it is water-soluble and is available in both enantiomeric forms.
As can be clearly seen in Fig. 1, the difference between isotropic and anisotropic spectra of L-proline is rather small, indicating that the desired small degree of order is induced by the LLC phase of 1, resulting in nicely resolved peaks (for extracted couplings see Table 1). Furthermore no signal originating from 1 is observed. We believe that this absence is due to the low concentration of 1 necessary to form the LLC phase and the excellent self-assembling properties of 1 that results in broadening of the resonances below detection limits.9 Thus it is possible to use β-peptidic LLC phases to align a water soluble small molecule and to obtain RDCs of the desired size.
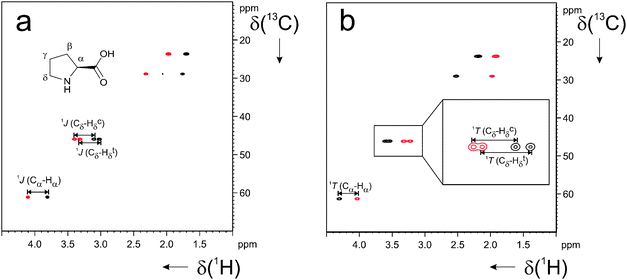 | ||
| Fig. 1 CLAP-HSQC spectrum12 of L-proline (ca. 7 mg) in (a) isotropic D2O solution and in (b) ca. 2.1% w/w LLC phase of β-peptide 1 in D2O. | ||
| L-Proline | D-Proline | ||||
|---|---|---|---|---|---|
| Atoms | J/ Hz | T/ Hz | D/ Hz b | T/ Hz | D/ Hz b,c |
| a The diastereotopic protons at the γ-position are isochronous in D2O. b The error of D is calculated as root mean square of T and J divided by 2 and is normalised for D-proline (see footnote c). c Normalisation was performed using the ratio of quadrupolar splittings of the solvent: DD-proline, norm = DD-proline. meas × 19.3/15.1. | |||||
| Cα–Hα | 148.4 ± 0.2 | 136.8 ± 0.5 | −5.8 ± 0.3 | 139.0 ± 0.7 | −6.0 ± 0.5 |
| Cδ–Hδc | 147.1 ± 0.3 | 138.8 ± 1.0 | −4.2 ± 0.5 | 140.4 ± 0.8 | −4.3 ± 0.5 |
| Cδ – Hδ t | 146.8 ± 0.2 | 158.1 ± 1.0 | 5.6 ± 0.5 | 157.5 ± 1.0 | 6.8 ± 0.6 |
| Cβ – Hβ c | 142.0 ± 0.5 | 133.5 ± 1.0 | −4.2 ± 0.6 | 132.3 ± 1.0 | −6.2 ± 0.7 |
| Cβ–Hβt | 134.5 ± 1.0 | 139.4 ± 1.0 | 2.4 ± 0.7 | 139.5 ± 1.0 | 3.2 ± 0.9 |
| Cγ – Hγ c /t a | 135.3 ± 0.2 | 135.5 ± 0.5 | 0.1 ± 0.3 | 133.4 ± 0.4 | −1.2 ± 0.3 |
To find out whether 1 is able to enantiodifferentiate, in addition to inducing the desired low degree of order, we prepared a sample of D-proline in a 1.7% w/w LLC phase of 1 in D2O (ΔνQ = 15.1 Hz) and looked for differences in the observed RDCs. A different concentration of 1, relative to the previous experiments, was chosen to probe for the expected concentration-dependence of induced order. As can be seen from the difference in quadrupolar splittings of the solvent in the two samples (19.3 Hz for 2.1% w/w vs. 15.1 Hz for 1.7% w/w), the expected concentration-dependence is indeed observed.
To be able to compare RDCs in the two samples, and to thus investigate enantiodiscrimination, we used the quadrupolar splitting of the solvent for scaling of RDCs as proposed previously.13 When comparing the RDCs for L- and D-proline in the LLC phase of 1 (Table 1) it can be seen that the values obtained for the two enantiomers are different (beyond experimental uncertainty) for 3 out of 6 couplings (bold entries in Table 1). This observation can be taken as a first indication that the two enantiomers are oriented differently, as has previously been shown for enantiomers of solutes oriented in α-peptidic poly-γ-benzyl-L-glutamate (PBLG) and gels based on naturally occurring triple helices (gelatin and collagen).14
We investigated whether the difference in chemical shift anisotropies (CSA) is large enough to lead to different signal sets when looking at scalemic mixtures. Such a difference was not observed (see ESI‡), which is consistent with our previous observations in high MW PBLG LLC phases.8 We believe that different chemical shifts could be observed if stronger orientation—leading to larger RDCs and larger CSA—were induced. This condition cannot be achieved with 1, however, because of limitations associated with physical properties (extremely high viscosity of the LLC phase at the concentrations that would be necessary).
Our results show that lyotropic liquid crystalline phases generated with β-peptides such as 1 can be used as alignment media. The degree of order induced is much lower than in LLC phases generated from the α-peptide PBLG, in which observed couplings frequently exceed 50–100 Hz, making the precise and accurate extraction of RDCs almost impossible. Both enantiomers of proline were oriented, and RDCs of suitable size were obtained. First indications towards enantiodiscrimination were observed, which could ultimately allow the determination of the absolute configuration of solutes.15 In the future we plan to take advantage of the fact that β-peptides are tunable mesogens, the properties of which can be modified via changing side chains (e.g., replacing cationic side chains with anionic side chains).10
C.M.T thanks Prof. M. Reggelin for his continuous support and the DFG (TH1115/3-1 and TH1115/5-1) and the Adolf Messer foundation for funding. We thank the BMRZ Frankfurt for measurement time at the 800 MHz NMR spectrometer. The work at UW-Madison was supported by the Nanoscale Science and Engineering Center (NSF DMR-0832760).
Notes and references
- T. D. W. Claridge, High Resolution NMR Techniques in Organic Chemistry, Elsevier, 2nd edn, 2009 Search PubMed.
- Reviews: (a) J. H. Prestegard, C. M. Bougault and A. I. Kishore, Chem. Rev., 2004, 104, 3519 CrossRef CAS; (b) J. R. Tolman and K. Ruan, Chem. Rev., 2006, 106, 1720 CrossRef CAS.
- Reviews: (a) C. M. Thiele, Eur. J. Org. Chem., 2008, 5673 CrossRef CAS and references therein; (b) G. Kummerlöwe and B. Luy, Annu. Rep. NMR Spectrosc., 2009, 68, 193 and references therein.
- (a) A. Saupe, Angew. Chem., Int. Ed. Engl., 1968, 7, 97 CrossRef CAS; (b) J. W. Emsley and J. C. Lindon, NMR Spectroscopy using liquid crystal solvents, Pergamon Press, Oxford, U.K., 1975 Search PubMed; (c) E. E. Burnell and C. A. de Lange, NMR of Ordered Liquids, Kluwer Academic Publishers, Dordrecht, 2003 Search PubMed; (d) N. Tjandra and A. Bax, Science, 1997, 278, 1111 CrossRef CAS.
- (a) B. Deloche and E. T. Samulski, Macromolecules, 1981, 14, 575 CrossRef CAS; (b) C. Gayathri, N. V. Tsarevsky and R. R. Gil, Chem.–Eur. J., 2010, 16, 3622 CrossRef CAS; (c) G. Kummerlöwe, E. F. McCord, S. F. Cheatham, S. Niss, R. W. Schnell and B. Luy, Chem.–Eur. J., 2010, 16, 7087.
- (a) P. W. Kuchel, B. E. Chapman, N. Müller, W. A. Bubb, D. J. Philp and A. M. Torres, J. Magn. Reson., 2006, 180, 256 CrossRef CAS; (b) G. Kummerlöwe, F. Habach, B. Laufer and B. Luy, Open Spectrosc. J., 2008, 2, 29 Search PubMed.
- Scaling of anisotropic NMR observables is also possible with variable angle sample spinning NMR: (a) J. Courtieu, J. P. Bayle and B. M. Fung, Prog. Nucl. Magn. Reson. Spectrosc., 1994, 26, 141 CrossRef CAS; (b) C. M. Thiele, Angew. Chem., Int. Ed., 2005, 44, 2787 CrossRef CAS.
- High MW PBLG has favourable orienting properties due to the reduced ccrit A. Marx and C. M. Thiele, Chem.–Eur. J., 2009, 15, 254 Search PubMed.
- M. M. Müller, M. A. Windsor, W. C. Pomerantz, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2009, 48, 922 CrossRef CAS.
- (a) W. C. Pomerantz, N. L. Abbott and S. H. Gellman, J. Am. Chem. Soc., 2006, 128, 8730 CrossRef; (b) W. C. Pomerantz, V. M. Yuwono, C. L. Pizzey, J. D. Hartgerink, N. L. Abbott and S. H. Gellman, Angew. Chem., Int. Ed., 2008, 47, 1241 CrossRef CAS; (c) W. C. Pomerantz, R. Drake, V. M. Yuwono, J. D. Hartgerink, N. L. Abbott, and S. H. Gellman, manuscript in preparation.
- (a) K. Czarniecka and E. T. Samulski, Mol. Cryst. Liq. Cryst., 1981, 63, 205–214 CAS; (b) A. Meddour, I. Canet, A. Loewenstein, J. M. Pechine and J. Courtieu, J. Am. Chem. Soc., 1994, 116, 9652 CrossRef CAS; (c) M. Sarfarti, P. Lesot, D. Merlet and J. Courtieu, Chem. Commun., 2000, 2069 RSC.
- A. Enthart, J. C. Freudenberger, J. Furrer, H. Kessler and B. Luy, J. Magn. Reson., 2008, 192, 314 CrossRef CAS.
- C. Canlet, D. Merlet, P. Lesot, A. Meddour, A. Loewenstein and J. Courtieu, Tetrahedron: Asymmetry, 2000, 11, 1911 CrossRef CAS.
- (a) A. Meddour, I. Canet, A. Loewenstein, J. M. Pechine and J. Courtieu, J. Am. Chem. Soc., 1994, 116, 9652 CrossRef CAS; (b) A. Marx, V. Schmidts and C. M. Thiele, Magn. Reson. Chem., 2009, 47, 734 CrossRef CAS; (c) Review: B. Luy, J. Indian Inst. Sci., 2010, 90, 119 Search PubMed and references therein.
- For a first example, in which this was achieved by comparison with a solute of known absolute configuration, see: L. Ziani, P. Lesot, A. Meddour and J. Courtieu, Chem. Commun., 2007, 4737 Search PubMed.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Spectrum of D-Proline in LLC phase, 2H spectra of both samples with exact sample compositions, spectra of scalemic mixture. See DOI: 10.1039/c0cc02123g |
| § Current address: University of Michigan, Ann Arbor, MI 48109, USA. |
| ¶ For sample preparation the corresponding amount of 1 (6–7 mg) was weighed into an Eppendorf tube, the appropriate amount of D2O was added, the Eppendorf tube was vortexed extensively, sealed using Parafilm® and put onto a shaker for several hours (at least 2 h). The sample was transferred into a 3 mm NMR tube using a syringe and its homogeneity and stability checked by 2H NMR. 7 mg of proline is added (at natural abundance, either as a solid or dissolved in D2O, no difference in stability of the phase was observed) and the sample is again vortexted extensively and its homogeneity etc. checked as above (for resulting ΔvQs, see text). RDCs are calculated from the difference between the line splittings in anisotropic and isotropic spectra and divided by 2 (T = J + 2D). All spectra (other than those of the scalemic mixture) were recorded on a Bruker DRX 500 NMR-spectrometer. |
| This journal is © The Royal Society of Chemistry 2011 |