Coarse-grained simulations of stretching entangled DNA using oscillating electric fields†‡
Richard S.
Graham
*a and
Ronald G.
Larson
b
aSchool of Mathematical Sciences, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: Richard.Graham@nottingham.ac.uk
bDepartment of Chemical Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA
First published on 8th September 2010
Abstract
DNA stretching in entangling media with oscillating electric fields is useful in DNA sequencing. We propose that stretching occurs around hairpin loops at the chain ends, caused by interactions with the medium. Brownian dynamics simulations show that this mechanism explains several experimental observations, including a resonance with varying field frequency.
DNA sequencing is essential to modern molecular biology and the need for evermore efficient and convenient sequencing methods is expected to grow as future applications are found.1 However, existing techniques require either bulk amounts of DNA2 or must sacrifice spatial resolution.3 An ideal technique would combine rapid, high-resolution sequencing from a small number of molecules. In this context, DNA stretching offers a promising new approach. If a DNA molecule can be stably elongated, the spatial positions of specific nucleotide sequences can more easily be found, either by selectively binding a fluorescent probe to a particular sequence4 or by splicing the chains with enzymes.5 The use of oscillating electric fields to stretch DNA6 is particularly attractive since it may be incorporated into practical, integrated DNA analysis devices.7,8 Studies of electrostretching DNA in entangling media such as cross-linked gels or neutral linear polymers are beginning to unearth some universal features:6,9–11 an optimum field frequency for stretching is found, suggesting a resonance effect with the molecular motion; and DNA are far more readily stretched in entangling media than in free solution,6 suggesting that the primary stretching mechanism is due to interactions with the entangling medium. Extrapolation of these results to situations of more practical relevance, such as longer DNA, requires a thorough understanding of the molecular basis for electrostretching. In this communication we use a recent molecular simulation for electrophoresis of DNA12 to model electrostretching and show that it is able to predict many of the observed experimental phenomena. The model is based on a generalisation of a modern coarse-grained model for bulk flows of entangled polymers13 and illustrates the potential for a unified molecular model applicable to both fields.
We postulate that DNA stretching results from transient hairpin shapes, formed at the trailing chain end (Fig. 1) around the fibres of the entangling medium. In this configuration chains are pulled from both ends (Fig. 1a) and subsequent reversal of the field allows stretching from the opposite chain end (Fig. 1b). Thus stretching is sustained with minimal centre of mass displacement. This suggests that optimum stretching occurs if the field reverses as the hairpin is broken, meaning the optimal field frequency is related to the hairpin lifetime. As biasing of the chain end motion by the field is critical to this mechanism, a detailed account of this process is essential.
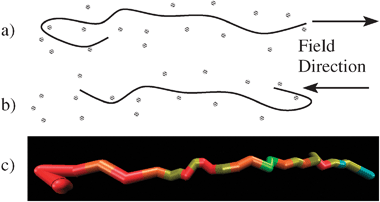 | ||
| Fig. 1 (a and b) Entangled DNA stretching about transient hairpins, formed on reversal of the field. (c) A simulated chain primitive path in a hairpin configuration. Colours represent the local stretch, with red being highly stretched through to light green being unstretched. The field is ε = 10, ωτe = 0.3. See also the ESI‡ for a related movie. | ||
Our simulation algorithm12 coarse-grains chains at the level of the entanglement spacing, taking the position of the entanglement points {Qi} and the number of monomers between each point {Ni} as the dynamic variables. The entanglements arise because the DNA chain cannot pass through the fibres of the entangling medium. The free energy of each strand has contributions from both the external electric field and the strand entropy, which is modelled as a non-Gaussian random walk. Gradients in the free energy drive both monomer sliding across the entanglement nodes and creation and destruction of entanglement junctions at the chain ends, which are resolved by a Brownian dynamics algorithm. In particular, this provides a detailed account of the influence of the electric field on the chain end dynamics. Key quantities in the model are the tube diameter a, which is related to the gel pore size, the number of Kuhn steps per entanglement segment Ne, the number of entanglement segments occupied in equilibrium Z and the relaxation time of an entanglement segment τe. Implicit time stepping is used to deal with the steep gradients in the chain free energy due to finite chain extensibility. Full details of this algorithm are provided elsewhere.12
The oscillating field is given by E(t) = E0sin(ωt), where t is time, E0 is the field amplitude and ω the frequency. A key modelling quantity is the dimensionless field 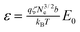 , where qζ is the effective charge per Kuhn step, b is the Kuhn step length and T is the temperature. The field strengths of ∼50–250 V cm−1 used by Kaji et al.10 for electrostretching correspond to ε ≈ 10–30 using literature parameters.12 Strictly, these large fields are beyond the range of the coarse-graining of our simulation algorithm, as they may excite fast processes on length scales below the tube diameter. However we correct for these effects in an approximate way, outlined below. The resulting model then establishes that the above stretching mechanism accounts for many features of stretching experiments. We correct the simulation algorithm for these high fields in two ways. High fields occasionally cause unrealistically long new entanglement segments to be created at the chain ends. We correct this by limiting to length of newly created segments to below some maximum Qmax. We take Qmax = 3.0, meaning that new segments must be less than 3 times the equilibrium length. The high fields also cause entanglement creation and destruction to be unphysically frequent. Thus we enforce that each newly created end segment must exist for a minimum time, of order of the relaxation time of the chain section in the dangling end. Upon creation or destruction of an entanglement segment, we forbid further entanglement renewal at that end for a period Δt = Se(Ns/Ne)2τe. Here Ns is the number of monomers in the end segment, (Ns/Ne)2τe is the timescale for the chain end section to fully explore its tube segment and Se is a parameter that controls the onset of this restriction. Throughout we take Se = 0.02, chosen to allow all but the most rapid entanglement renewal events.
, where qζ is the effective charge per Kuhn step, b is the Kuhn step length and T is the temperature. The field strengths of ∼50–250 V cm−1 used by Kaji et al.10 for electrostretching correspond to ε ≈ 10–30 using literature parameters.12 Strictly, these large fields are beyond the range of the coarse-graining of our simulation algorithm, as they may excite fast processes on length scales below the tube diameter. However we correct for these effects in an approximate way, outlined below. The resulting model then establishes that the above stretching mechanism accounts for many features of stretching experiments. We correct the simulation algorithm for these high fields in two ways. High fields occasionally cause unrealistically long new entanglement segments to be created at the chain ends. We correct this by limiting to length of newly created segments to below some maximum Qmax. We take Qmax = 3.0, meaning that new segments must be less than 3 times the equilibrium length. The high fields also cause entanglement creation and destruction to be unphysically frequent. Thus we enforce that each newly created end segment must exist for a minimum time, of order of the relaxation time of the chain section in the dangling end. Upon creation or destruction of an entanglement segment, we forbid further entanglement renewal at that end for a period Δt = Se(Ns/Ne)2τe. Here Ns is the number of monomers in the end segment, (Ns/Ne)2τe is the timescale for the chain end section to fully explore its tube segment and Se is a parameter that controls the onset of this restriction. Throughout we take Se = 0.02, chosen to allow all but the most rapid entanglement renewal events.
We base our model parameters on the experimental conditions of Kaji et al.10 and the literature parameters used to successfully model constant field electrophoresis with this simulation algorithm.12 Thus a 165 kbp DNA chain in a 0.5–3% agarose gel corresponds to Z = 20 entanglements, with Ne = 100. We use a field amplitude of ε = 10 and simulate the effect a wide range of frequencies on DNA stretching, with the results summarised in Fig. 2. The length along the field direction x is defined as the distance, in the field direction, between the two furthest apart chain segments. The primitive path is the sum over the lengths of all entanglement segments, regardless of their direction, and it corresponds to the overall polymer stretch. Also shown is a measure of the polymer's displacement, given by the mean of the absolute distance moved by the chain's centre of mass during a half cycle of the field. At low frequency this average displacement is substantially longer than the chain length and is excluded from Fig. 2. Similarly to experiments,6,10,11 we found that the evolution towards the stretched state is hampered by the transient formation of long-lived looped configuration, particularly at high frequencies. Experimentally, this is avoided through carefully increasing the field frequency to align the chain prior to stretching.11 To mimic this “frequency annealing”, we performed each simulation at a fixed frequency but began with a fully aligned but unstretched chain. Our results are averages over several independent simulations, each of at least 100 field cycles. The first few cycles of each run contain memory of the starting configuration so are excluded from the averaging.
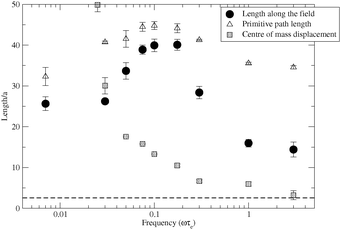 | ||
| Fig. 2 Simulations of chain stretch and centre of mass displacement. The dashed line is the equilibrium chain length along the field. | ||
Our stretching results have many similarities to electrostretching experiments.6,10,11 The stretching in the field direction, as shown by 〈x〉, displays a clear optimum frequency, at which the chains are stretched to roughly double their low frequency value. Further increase in frequency leads to a strong reduction in 〈x〉. The optimum stretching frequency is correlated to the centre of mass displacement. At low frequencies the centre of mass displacement exceeds 〈x〉 and this suppresses the stretching. Around the optimum frequency there is a sharp decrease in the centre of mass displacement to substantially less than the 〈x〉, indicating strong chain localisation as well as stretching. This localisation persists to the higher frequencies even though the stretching is suppressed once more. Exactly this connection between the centre of mass displacement and the three frequency regimes was seen experimentally.10 The simulations also reveal further details of the stretching process. In Fig. 2 the primitive path stretching is weak at low frequencies. Here, the chains are aligned in the field direction but remain mostly unstretched, with occasional cycles through elongated configurations, similarly to constant field electrophoresis.6 Beyond the optimum frequency, the primitive path is strongly elongated but this does not translate into extension of 〈x〉. This indicates that chains stretch, but are repeatedly doubled over in a zig-zag configuration with modest 〈x〉. At these high frequencies the premature reversal of the electric field, before the chain is fully stretched around the hair-pin, causes new entanglement segments to be incoherently placed. All of these results are consistent with the stretching mechanism proposed in Fig. 1. As further support, Fig. 1c shows a typical hairpin configuration from the simulations. Such configurations are substantially more common in the stretching regime. A movie in the ESI‡ shows the mechanism in effect during a stretching simulation for ε = 10 and ωτe = 0.3. The optimum frequency is set by the hairpin lifetime. For optimum stretching the field must reverse just as the hairpin is broken, allowing another to form at the opposite end. This hairpin lifetime depends on the chain tension, force exerted by the field on the chain and total chain drag and so cannot be simply related to any molecular relaxation time. Understanding the dependence of the optimum frequency on field strength, DNA length and gel concentration will be essential in extending this modelling to practical applications.
In summary, we have proposed a mechanism for DNA stretching under oscillating fields, which is dominated by entanglement interactions between the DNA and surrounding medium (Fig. 1). This mechanism relies on transient hairpin configurations at the chain ends, which occur on reversal of the field. We accounted for the large electric fields used in electrostretching through an approximate modification of a Brownian dynamics simulation algorithm for DNA electrophoresis. The resulting model predicts many experimental features, including an optimal frequency for stretching and a correlation between this optimal frequency and the chain centre of mass displacement. The much higher spatial resolution of the simulations compared to experiments elucidates details of the stretching mechanism. For example, the role of transient hair-pins is clearly seen in the chain configurations during stretching (see Fig. 1c and the ESI‡). Also, the simulations show that at high frequencies the primitive path is stretched but that individual segments are incoherently placed so stretching in the field direction is modest, consistent with our proposed mechanism.
From our results it is clear that electrostretching is sensitive to molecular processes that occur on length scales both above and below the tube diameter. Therefore an improved electrostretching model would need a more detailed picture of the interaction of the chains with the entangling medium at these fine length scales. Models that resolve the cross over from sub-tube diameter to entangled dynamics exist from the modelling of linear rheology of synthetic polymers.14 However, this model's algorithm of entanglement creation and destruction at the chain ends needs to be generalised to non-equilibrium situations for electrophoresis modelling to be possible.
Comparing electrostretching experiments with molecular models improves both the practical and fundamental understanding of this important process. DNA also has some interesting advantages as a model polymer. Insight gained from single model experiments can be incorporated into rheological models of entangled polymers.15 The correspondence in the dynamics between synthetic polymers and DNA in the entangled regime has been established through bulk measurement and single molecule visualisation for entangled DNA solutions.16 This work shows the potential for a unified model of non-equilibrium polymer dynamics, covering both fields.
Notes and references
- M. A. Burns, Science, 2002, 296, 1818–1819 CrossRef CAS.
- D. T. Burke, G. F. Carle and M. V. Olson, Science, 1987, 236, 806 CAS.
- P. R. Langer-Safer, M. Levine and D. C. Ward, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 4381 CrossRef CAS.
- H. Oana, M. Ueda and M. Washizu, Biochem. Biophys. Res. Commun., 1999, 265, 140 CrossRef CAS.
- D. C. Schwartz, X. J. Li, L. I. Hernandez, S. P. Ramnarain, E. J. Huff and Y. K. Wang, Science, 1993, 262, 110 CrossRef CAS.
- M. Ueda, J. Biochem. Biophys. Methods, 1999, 41, 153 CrossRef CAS.
- M. A. Burns, B. N. Johnson, S. N. Brahmasandra, K. Handique, J. R. Webster, M. Krishnan, T. S. Sammarco, P. M. Man, D. Jones, D. Heldsinger, C. H. Mastrangelo and D. T. Burke, Science, 1998, 282, 484 CrossRef CAS.
- V. Namasivayam, R. G. Larson, D. T. Burke and M. A. Burns, Anal. Chem., 2002, 74, 3378 CrossRef CAS.
- M. Ueda, K. Yoshikawa and M. Doi, Polym. J. (Tokyo), 1997, 29, 1040 Search PubMed.
- N. Kaji, M. Ueda and Y. Baba, Biophys. J., 2002, 82, 335 CrossRef CAS.
- N. Kaji, M. Ueda and Y. Baba, Appl. Phys. Lett., 2003, 83, 3413 CrossRef CAS.
- R. S. Graham and R. G. Larson, Macromolecules, 2007, 40, 366 CrossRef CAS.
- J. D. Schieber, J. Neergaard and S. Gupta, J. Rheol. (N. Y.), 2003, 47, 213 Search PubMed.
- A. E. Likhtman, Macromolecules, 2005, 38, 6128 CrossRef CAS.
- T. C. B. McLeish, Adv. Phys., 2002, 51, 1379 CrossRef CAS.
- R. E. Teixeira, A. K. Dambal, D. H. Richter, E. S. G. Shaqfeh and S. Chu, Macromolecules, 2007, 40, 2461 CrossRef.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Movie of the simulated stretching. See DOI: 10.1039/c0cc02090g |
| This journal is © The Royal Society of Chemistry 2011 |