Synthetically accessible, tunable, low-molecular-weight oligopeptide organogelators†‡
Cecile
Lagadec
and
David K.
Smith
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: dks3@york.ac.uk
First published on 25th August 2010
Abstract
We report a synthetically simple approach to effective and tunable low molecular weight gelators based on amine-initiated oligomerisation of an amino acidN-carboxyanhydride.
Self-assembling nanostructured gels, in which non-covalent interactions between molecular building blocks assemble a nanoscale ‘solid-like network’ suspended within a ‘liquid-like’ solvent phase, are of intense interest.1 The components used to assemble such materials can either be small molecules or polymers. However, there has been relatively little overlap between researchers in these distinct areas.2Peptides are capable of mutually complementary hydrogen bond interactions, and there has therefore been considerable interest in using peptides to assemble gels.3 The diversity of amino acid side chains, combined with their biocompatibility, opens a variety of applications.4 Low molecular weight peptides, often incorporating hydrophobic amino acids, have been widely employed, as organogelators.5 The peptides aggregate through hydrogen bonding whilst the apolar groups maintain the solubility in the organic solvent and avoid precipitation. Polymeric peptides have also been assembled into physical organogels. For example, poly(γ-benzyl-L-glutamate) forms organogels in toluene due to aggregation between polypeptide helices.6Polypeptides have been incorporated into block co-polymers which can form gels at low loadings.7Amino acids have also been attached to polymers, either as side chains, or on the termini, in order to provide the polymers with greater propensity to self-assemble into gels.8 For some time, we have investigated gelators which are intermediate between low and high molecular weight, working extensively with dendritic peptides,9 and gaining a good understanding of their assembly.10 However, dendrimers are relatively complex and require multi-step syntheses. We therefore became interested in developing alternative approaches to gelator structures of intermediate size. The preliminary results are reported here. We decided to apply ring opening oligomerisation using an amino acidN-carboxyanhydride to yield relatively small oligopeptides.11 Clearly, the products will not be as well-defined as traditional low molecular weight gelators, but the ease of synthesis is highly attractive. We were interested to know whether the polydisperse crude products from this type of reaction could still support gelation.
To demonstrate this approach we used Nε-(benzyloxycarbonyl)-L-lysine N-carboxyanhydride ( NCA), as we have previously shown that well-defined peptides constructed from Z-protected lysine can form effective gels.12 Compound NCA was synthesised in 84% yield viacyclisation of Nε-(benzyloxycarbonyl)-L-lysine with triphosgene under argon in anhydrous THF (see ESI†).13 We then explored the reaction of NCA using a range of amines which can initiate ring opening oligomerisation of a peptide chain (Scheme 1). A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶6 initiator∶ NCA ratio was employed to target relatively short peptide oligomers. Initiators 1–6 (Scheme 1, for other initiators see ESI†) were investigated using a parallel reaction station, with the crude products being dissolved in a minimum of DMF and the desired oligomers precipitated by the addition of diethyl ether.
∶6 initiator∶ NCA ratio was employed to target relatively short peptide oligomers. Initiators 1–6 (Scheme 1, for other initiators see ESI†) were investigated using a parallel reaction station, with the crude products being dissolved in a minimum of DMF and the desired oligomers precipitated by the addition of diethyl ether.
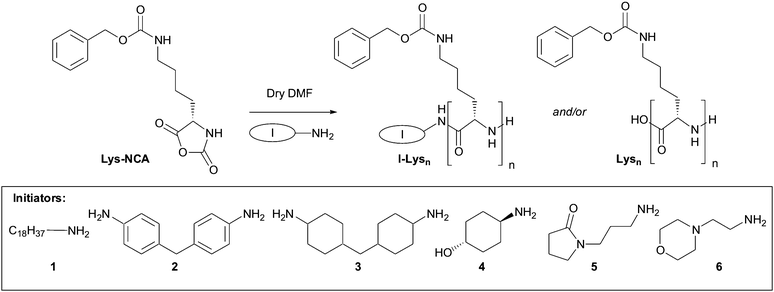 | ||
| Scheme 1 Amine-initiated oligomerisation of Z-protected lysine N-carboxyanhydride. Initiators 1–6 were tested in this reaction. | ||
Electrospray mass spectrometry (ESMS) showed that, in most cases, the crude solid products formed from initiators 1–6 had succeeded in growing an oligolysine chain to give the desired I–Lysn. Typically, between two and six lysine units were attached to the initiator (Table 1). However, the products obtained in some cases also had ESMS peaks for unattached oligo-lysine ( Lysnnn, Fig. S1, ESI†)—oligomerisation was initiated with adventitious water, possibly because these initiators were difficult to dry fully. Two initiators ( 1 and 4) did not produce any unattached oligo-lysine, but initiator 6 mainly gave Lysnnn, with a smaller amount of I–Lysn.§
| 1 (%) | 2 (%) | 3 (%) | 4 (%) | 5 (%) | 6 (%) | |
|---|---|---|---|---|---|---|
| Lys44 | — | 39 | 18 | — | 16 | 88 |
| Lys55 | — | 39 | — | — | 16 | 100 |
| Lys66 | — | 18 | — | — | 11 | 55 |
| Lys77 | — | 6 | — | — | 3 | 24 |
| Lys88 | — | — | — | — | — | 6 |
| I– Lys 2 | 100 | — | — | 100 | 100 | 9 |
| I– Lys 3 | 94 | 67 | 58 | 60 | 73 | 9 |
| I– Lys 4 | 53 | 100 | 100 | 20 | 40 | 4 |
| I– Lys 5 | 56 | 56 | 33 | 5 | 3 | 2 |
| I– Lys 6 | 22 | 22 | 33 | — | 3 | 2 |
| I– Lys 7 | 12 | 11 | 42 | — | — | — |
| I– Lys 8 | 44 | 6 | — | — | — | — |
The gelation of the crude oligopeptide products was then studied in a range of solvents. Pleasingly, most compounds formed gels in 1,2-dichlorobenzene (DCB, Table 2). The exception to this was 6-Lys—interestingly, the product which was primarily unattached Lysnnn—suggesting that this peptide does not assemble into effective gels, and may even suppress gelation. Only 1–Lys formed gels in toluene (TOL), and only 3–Lys gelated trichloroethylene (TCE), with 1–Lys forming a viscous liquid in TCE. This demonstrates that the choice of initiator directly impacts on gelation ability.
| Sola | α | β | π* | 1–Lys | 2–Lys | 3–Lys | 4–Lys | 5–Lys | 6–Lys |
|---|---|---|---|---|---|---|---|---|---|
| a DEC (decane); MES (mesitylene); TCE (trichloroethylene); TOL (toluene); DCB (1,2-dichlorobenzene); CAN (acetonitrile); NM (nitromethane). | |||||||||
| DEC | 0.00 | 0.00 | 0.30 | Ins | Ins | Ins | Ins | Ins | Ins |
| MES | 0.00 | 0.13 | 0.41 | Ins | Ins | Ins | Ins | Ins | Ins |
| TOL | 0.00 | 0.11 | 0.49 | G | Ins | Ins | Ins | Ins | Ins |
| TCE | 0.00 | 0.50 | 0.53 | V | Ins | G | Ins | Ins | Ins |
| DCB | 0.00 | 0.30 | 0.80 | G | G | G | G | G | V |
| ACN | 0.19 | 0.40 | 0.75 | Ins | Ins | Ins | Ins | Ins | Ins |
| NM | 0.22 | 0.06 | 0.85 | Ins | Ins | Ins | Ins | Ins | Ins |
Considering the solvent Kamlet–Taft parameters (Table 2),14gelation only occurred if the α parameter was zero, i.e., hydrogen bond donor solvents such as acetonitrile (ACN) and nitromethane (NM) disrupt gelation. Of those solvents with an α parameter equal to zero, only DCB and to some extent TCE and TOL supported gelation, whereas decane (DEC) and mesitylene (MES) did not. It is likely that gelation occurred best in DCB because it has quite high π* (polarisability) and β (hydrogen bond acceptor) values—which are required in order to solubilise the moderately polar oligopeptide. If the π* and β values are too low (e.g.DEC and MES), then the oligopeptides will not dissolve, hence preventing gelation from taking place. We reason that TCE and TOL are somewhat intermediate in terms of their π* and β values and therefore only support gels for some of the molecular frameworks.
We investigated 1–Lys in more detail and monitored the gelation ability of this compound as a function of concentration, using a vial inversion method to record Tgel values (Fig. 1). The Tgel value rose to >120 °C at approximately 3 mg mL−1 (0.3 wt/vol%) in DCB. The minimum gelation concentration (MGC) at room temperature was ca. 0.15 wt/vol%. This is very impressive gelation at such low loading and compares very favourably to other gels. Typically, peptide organogels3,5,9,10 have MGC values of >0.2 wt/vol% at room temperature—often significantly more. Importantly, this clearly demonstrates that the polydisperse nature of compound 1–Lys does not inhibit its ability to act as an effective organogelator. Compounds 2–Lys and 3–Lys were also investigated (Fig. 1). These gelators also had low MGC values—the MGC of 3–Lys was ca. 0.15 wt/vol%, whereas 2–Lys was less effective (0.65 wt/vol%). However, these gelators had lower Tgel values than 1–Lys. Gelator 3–Lys had a higher Tgel value (ca. 40 °C) than 2–Lys (ca. 35 °C). It is possible that 2–Lys is less effective due to structural features of the initiator, although this may be a consequence of 2–Lys containing more unattached Lysnnn, which is not an effective gelator—and may even inhibit the gelation process somewhat. Interestingly, 4–Lys (see below) also gave less thermally stable gels than 1–Lys, even though it contains no Lysnnn. This demonstrates that the chemical structure of the initiator plays an active role in controlling the thermal properties of the gel. It is possible that the apolarity of initiator 1 enhances gel stability, for example through van der Waals interactions or solubility modification.12
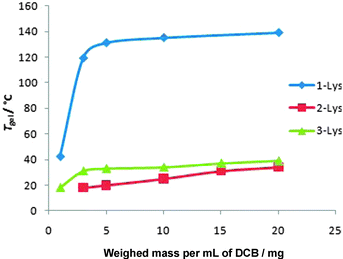 | ||
| Fig. 1 Thermal behaviour of gels formed from 1–Lys, 2–Lys and 3–Lys in 1,2-dichlorobenzene as solvent. | ||
The influence of oligomerisation was studied in more detail using initiator 4 with different 4∶ NCA molar ratios—1∶2, 1∶4 and 1∶10. The 1∶2 ratio led to a product which could not be precipitated—it was assumed that any oligomers obtained were too short. The other reactions produced solid products. ESMS indicated that the compound formed from the 1∶4 ratio reaction ( 4–Lys-A) had 2 or 3 lysines linked to the initiator, with the major peak being for 2 lysines and with no Lysnnn being observed (Fig. S3, ESI†). On the other hand, the crude product from the 1∶10 reaction ( 4–Lys-B) exhibited ESMS peaks for longer oligomers having 2–5 lysine units attached to the initiator, with the major peaks for 3 and 4 lysine repeat units—however, some peaks were also observed for Lysnnn (Fig. S4, ESI†). Clearly, when larger amounts of NCA were used, the oligomer chain length increases, but so does the chance of oligomerisation initiated by adventitious water. Compound 4–Lys-A formed moderately stable gels with a maximum Tgel value around 35 °C (Fig. 2). Compound 4–Lys-B had a lower thermal stability (<25 °C). Clearly, the presence of longer oligomers does not benefit gel stability—indeed, relatively short oligo-lysines attached to the initiator appear sufficient to enable efficient self-assembly and avoid the formation of Lysnnn. Scanning electron microscopy (SEM) performed on the xerogels indicated that although both these gels had a fibrillar nature (Fig. S5 and S6, ESI†), similar to related low molecular weight well-defined peptide gels,104–Lys-A had a more nanostructured appearance.
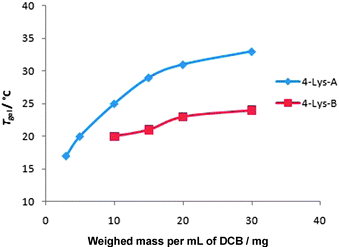 | ||
| Fig. 2 Thermal behaviour of gels formed from 4–Lys-A and 4–Lys-B in 1,2-dichlorobenzene as solvent. | ||
This communication demonstrates that oligomerisation of amino acid NCA derivatives is a simple approach to generating oligomers, which even in crude form, can be excellent organogelators. In this case, the use of initiator 1 is optimal. Structurally perfect small molecules are not, therefore, a necessary pre-requisite for gelation. Features such as choice of initiator, and length of oligomer control gel behaviour, making these materials highly tunable. Future work will focus on high-throughput screening of further oligopeptides, minimising water-initiated oligomers, and exploiting potential applications. For example, we anticipate that modifying the amino acid, will give rise to a versatile class of synthetically simple nanomaterials.
We acknowledge EPSRC and Givaudan for funding.
Notes and references
- Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. P. Terech and R. G. Weiss, Springer, Dordrecht, 2006 Search PubMed.
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2010, 39, 455–463 RSC.
- F. Fages, F. Vögtle and M. Zinic, Top. Curr. Chem., 2005, 256, 77–131 CAS.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- For selected examples: (a) J.-H. Fuhrhop, D. Spiroski and C. Boettcher, J. Am. Chem. Soc., 1993, 115, 1600–1601 CrossRef CAS; (b) K. Hanabusa, R. Tanaka, M. Suzuki, M. Kimura and H. Shirai, Adv. Mater., 1997, 9, 1095–1097 CrossRef CAS; (c) S. Bhattacharya and S. N. G. Acharya, Chem. Mater., 1999, 11, 3121–3132 CrossRef CAS; (d) B. Escuder, S. Martí and J. F. Miravet, Langmuir, 2005, 21, 6776–6787 CrossRef CAS; (e) A. Friggeri, C. van der Pol, K. J. C. van Bommel, A. Heeres, M. C. A. Stuart, B. L. Feringa and J. van Esch, Chem.–Eur. J., 2005, 11, 5353–5361 CrossRef CAS; (f) I. A. Coates, A. R. Hirst and D. K. Smith, J. Org. Chem., 2007, 72, 3937–3940 CrossRef CAS; (g) H.-F. Chow, J. Zhang, C.-M. Lo, S.-Y. Cheung and K.-W. Wong, Tetrahedron, 2007, 63, 363–373 CrossRef CAS; (h) D. Bardelang, M. B. Zaman, I. L. Moudrakovski, S. Pawsey, J. C. Margeson, D. Wang, X. Wu, J. A. Ripmeester, C. I. Ratcliffe and K. Yu, Adv. Mater., 2008, 20, 4517–4520 CrossRef CAS; (i) A. Banerjee, G. Palui and A. Banerjee, Soft Matter, 2008, 4, 1430–1437 RSC; (j) V. Lozano, R. Hernandez, C. Mijangos and M. J. Perez-Perez, Org. Biomol. Chem., 2009, 7, 364–369 RSC.
- (a) T. Korenaga, H. Oikawa and H. Nakanishi, J. Macromol. Sci. Phys., 1997, 36, 487–501 CrossRef; (b) R. Tadmor, R. L. Khalfin and Y. Cohen, Langmuir, 2002, 18, 7146–7150 CrossRef CAS.
- (a) K. T. Kim, C. Park, G. W. M. van der Meulen, D. A. Rider, C. Kim, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2005, 44, 7964–7968 CrossRef CAS; (b) M. I. Gibson and N. R. Cameron, Angew. Chem., Int. Ed., 2008, 47, 5160–5162 CrossRef CAS.
- (a) H. Ihara, M. Takafuji, T. Sakurai, M. Katsumoto, N. Oshijima, T. Shirosaki and H. Hachisoko, Org. Biomol. Chem., 2003, 1, 3004–3006 RSC; (b) S. Okabe, K. Ando, K. Hanabusa and M. Shibayama, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 1841–1848 CrossRef CAS; (c) M. Suzuki, C. Setoguchi, H. Shirai and K. Hanabusa, Chem.–Eur. J., 2007, 13, 8193–8200 CrossRef CAS; (d) J. Hentschel and H. G. Borner, J. Am. Chem. Soc., 2006, 128, 14142–14149 CrossRef CAS.
- D. K. Smith, Adv. Mater., 2006, 18, 2773–2778 CrossRef CAS.
- (a) B. Huang, A. R. Hirst, D. K. Smith, V. Castelletto and I. W. Hamley, J. Am. Chem. Soc., 2005, 127, 7130–7139 CrossRef CAS; (b) A. R. Hirst, V. Castelletto, I. W. Hamley and D. K. Smith, Chem.–Eur. J., 2007, 13, 2180–2188 CrossRef CAS; (c) A. R. Hirst, D. K. Smith and J. P. Harrington, Chem.–Eur. J., 2005, 11, 6552–6559 CrossRef CAS; (d) A. R. Hirst and D. K. Smith, Org. Biomol. Chem., 2004, 2, 2965–2971 RSC.
- (a) J. Rodriguez-Hernandez, M. Gatti and H.-A. Klok, Biomacromolecules, 2003, 4, 249–258 CrossRef CAS; (b) I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944–2945 RSC; (c) T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS.
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS.
- R. Wilder and S. Mobashery, J. Org. Chem., 1992, 57, 2755–2756 CrossRef CAS.
- (a) M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS; (b) C. Laurence, P. Nicolet, M. T. Dalati, J. L. M. Abboud and R. Notario, J. Phys. Chem., 1994, 98, 5807–5816 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and assay procedures, sample mass spectra, SEM images and full MS characterisation data. See DOI: 10.1039/c0cc01449d |
| ‡ This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| § MS peak height does not necessarily correlate directly with product composition. However, for comparison of structurally related compounds it provides useful insight into product composition. For diamines, it is hard to say whether the growth of the oligomer occurs on both amines simultaneously or only on one. |
| This journal is © The Royal Society of Chemistry 2011 |