Self-regeneration of a silylium ion catalyst in carbonyl reduction†‡
Kristine
Müther
and
Martin
Oestreich
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität, Corrensstrasse 40, D-48149 Münster, Germany. E-mail: martin.oestreich@uni-muenster.de; Fax: +49 (0)25183-36501; Tel: +49 (0)25183-33271
First published on 25th August 2010
Abstract
The silylium ion-catalysed reduction of carbonyl compounds to the alcohol oxidation level is accomplished by exploiting the unique reactivity of a ferrocenyl-substituted silane.
The crux of silylium ion catalysis is to keep the extremely potent Lewis acid, that is the trivalent silicon cation1 itself, alive. Strongly binding Lewis bases must be scrupulously avoided, and any Lewis pair formation must be reversible. There are a few examples of silylium ion-catalysed processes, from which the silicon cation emerges indeed unchanged,2 but there are also efficient catalytic reactions involving covalent bond formation to the silicon atom of the silylium ion. To maintain turnover in those, a silylium ion must be regenerated from a suitable precursor in a subsequent step. That vital self-regeneration was established by Lambert et al. in a silylium ion-catalysed alkene hydrosilylation.3 A more prominent example is the silylium ion-catalysed C–F bond activation recently disclosed by Ozerov et al., a formal Si–H/C–F metathesis.4 Both reactions, while appearing unrelated, share however the regeneration of the solvent-stabilised silylium ion catalyst, namely the hydride abstraction from a silane5 by an intermediate carbenium ion.6
The situation is different in the silylium ion-catalysed reduction of carbonyl compounds,7 resulting in deoxygenation rather than reduction to the alcohol oxidation level. Kira et al. had investigated that unexpected outcome almost two decades ago,8 and an investigation by Piers et al. later corroborated the suggested mechanism (Scheme 1).9 The catalysis commences with the formation of silyloxycarbenium (or silylcarboxonium) ion 3 through coordination of the Lewis basic carbonyl oxygen of 2 to donor-stabilised silylium ion 1. The hydride affinity of 3 is not as pronounced as that of the previously discussed carbenium ions,10 and hydride abstraction from silane 4 will be less facile. Piers et al. had reasoned though that hydride transfer is likely to be assisted by the oxygen atom in 3,9 proceeding through tentative transition state 5 (3 → 6). Disiloxane 7 is a good leaving group, and oxonium ion 6 dissociates to produce carbenium ion 8, which in turn is sufficiently reactive to directly regenerate silylium ion 1 upon reaction with 4 (8 → 9). The net deoxygenation (2 → 9) was experimentally verified at room temperature for benzophenone8 and acetophenone9 as substrates.11
![The Kira–Piers mechanism of the silylium ion-catalysed deoxygenation of carbonyl compounds (Do = donor and R = Et). Counteranion [BAr4]− is omitted for clarity (Ar = 3,5-bis(trifluoromethyl)phenyl8 or pentafluorophenyl9).](/image/article/2011/CC/c0cc02139c/c0cc02139c-s1.gif) | ||
| Scheme 1 The Kira–Piers mechanism of the silylium ion-catalysed deoxygenation of carbonyl compounds (Do = donor and R = Et). Counteranion [BAr4]− is omitted for clarity (Ar = 3,5-bis(trifluoromethyl)phenyl8 or pentafluorophenyl9). | ||
Aside from the hydride affinity of an intermediate silylcarboxonium ion, the hydride donor strength of the silane is equally critical. Our laboratory recently introduced ferrocene-stabilised silylium ion 11,12,13 which was accessed by an exceptionally rapid Bartlett–Condon–Schneider hydride transfer14 (10 → 11, Scheme 2).
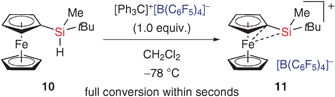
We then anticipated that the unusual hydridic character of 10 could allow for the reduction of silylcarboxonium ion 12 without concomitant oxonium ion formation (12 → 13, Scheme 3). Both the stabilisation of the trivalent silicon cation through the ferrocenyl substituent and the steric bulk of the reaction partners might account for immediate regeneration of “free” 11. In this communication, we report the silylium ion-catalysed reduction of carbonyl compounds, which hinges upon the self-regeneration of a catalytically active silicon cation by that mechanism (2 → 13, Scheme 3).
![Proposed silylium ion-catalysed reduction of carbonyl compounds. Counteranion [B(C6F5)4]− is omitted for clarity.](/image/article/2011/CC/c0cc02139c/c0cc02139c-s3.gif) | ||
| Scheme 3 Proposed silylium ion-catalysed reduction of carbonyl compounds. Counteranion [B(C6F5)4]− is omitted for clarity. | ||
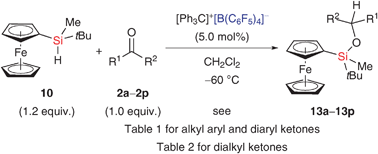
In light of the deoxygenation observed for aryl-substituted ketones (vide supra),8,9 we began our screening using these carbonyl compounds. Catalyst 11 was generated in CH2Cl2 at −60 °C from [Ph3C]+[B(C6F5)4]− (5.0 mol%) and a slight excess of 10 prior to addition of the reactants (Scheme 4).§ The reaction of acetophenone was smooth, affording the silicon ether in good yield (2a → 13a, Table 1, entry 1). No defunctionalisation was detected. Conversely, benzophenone was not participating in the catalysis at all (2b, Table 1, entry 2). This rather unexpected result might be explained by the relative hydride affinities of silylcarboxonium ions 12a and 12b (cf.Scheme 3): two phenyl groups (as in 12b) lend more stabilization to such intermediates than just one (as in 12a). The same rationale applies to naphthyl- and other electron-rich phenyl-substituted ketones (2c–2f, Table 1, entries 3–6).
| Entry | Carbonyl compound 2 | Silicon ether 13 | ||||
|---|---|---|---|---|---|---|
| No. | R1 | R2 | No. | drb | Yieldc (%) | |
| a All reactions were conducted using [Ph3C]+[B(C6F5)4]− (5.0 mol%) to generate 11, and silane (1.2 equiv.) and carbonyl compound (1.0 equiv.) with a ketone concentration of 0.2 M in CH2Cl2 at −60 °C. Reactions were terminated after 2½ h. b Diastereomeric ratio determined by GLC analysis prior to purification. c Combined yield of analytically pure mixture of diastereomers after flash chromatography. d Details are discussed in the text (Scheme 5). e Poor reactivity also due to steric factors. | ||||||
| 1 | 2a | Ph | Me | 13a | 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶49 ∶49 |
82 |
| 2 | 2b | Ph | Ph | 13b | — | No reaction |
| 3 | 2c | 2-C10H7 | Me | 13c | — | Tracesd |
| 4 | 2d | 1-C10H7 | Me | 13d | — | No reactione |
| 5 | 2e | 4-MeOC6H4 | Me | 13e | — | No reaction |
| 6 | 2f | 4-MeC6H4 | Me | 13f | 52∶48 |
10d |
| 7 | 2g | 2-ClC6H4 | Me | 13g | 78∶22 |
97 |
| 8 | 2h | C6F5 | Me | 13h | 52∶48 |
85 |
| 9 | 2i | Ph | Et | 13i | 78∶22 |
82 |
Only 2g–2i displayed the reactivity of 2a (2g–2i → 13g–13i, Table 1, entries 7–9), and the slightly more hindered substrates 2g and 2i showed a low level of diastereocontrol. The lack of facial selectivity might be interpreted in support of the hypothesis that silicon-to-carbon hydride transfer passes through an acyclic transition state (12 → 13, Scheme 3),15 not involving the oxygen atom (cf.4 → 5 → 6, Scheme 1).
The poor reactivity of ketones 2b–2f might be overcome at elevated reaction temperatures. The upper temperature limit of −30 °C is set by the chemical stability of silylium ion 11 in CH2Cl2.12 However, even an increase from −60 °C to −45 °C partially resulted in ipsoalkylation of the ferrocene in several cases (10 → 14, Scheme 5). We further analyzed this finding in the reduction of acetophenone at −45 °C: the silicon ether formed in the carbonyl-to-hydroxy reduction (2a → 13a) was readily converted into the alkylated ferrocene in 55% isolated yield (13a → 14a). Treatment of an independently prepared sample of 13a under the standard reaction conditions at −45 °C afforded 14a in 38% isolated yield. This set of data indicates to us that reduction occurs at higher reaction temperatures, followed by carbenium ion formation (cf.Scheme 1). The thus formed strong electrophile is then attacked by any of the present silylated ferrocenes, e.g., 13 or (tBuFcMeSi)2O (Fc = ferrocenyl), to undergo a Friedel–Crafts reaction.
![Deoxygenation—Friedel–Crafts sequence {[Ph3C]+[B(C6F5)4]− (5.0 mol%) was used to initiate the reaction}.](/image/article/2011/CC/c0cc02139c/c0cc02139c-s5.gif) | ||
| Scheme 5 Deoxygenation—Friedel–Crafts sequence {[Ph3C]+[B(C6F5)4]− (5.0 mol%) was used to initiate the reaction}. | ||
These experiments clearly reveal that the stability or, more precisely, the hydride affinity of the intermediate silylcarboxonium ion profoundly effects this catalysis. Electron-rich 4-MeOC6H4 (as in 2e) is deactivating, electron-poor C6F5 (as in 2h) is activating (Table 1, entries 5 and 8). The majority of aryl-substituted substrates forms unreactive 12 (Scheme 3 and Table 1). Based on this understanding, we expected 12 derived from purely alkyl-substituted carbonyls to be less stable and more reactive, and this is indeed the case. All dialkyl ketones surveyed performed very well (2j–2p → 13j–13p, Table 2). Diastereoselectivity was again low (vide supra).15
| Entry | Carbonyl compound 2 | Silicon ether 13 | ||||
|---|---|---|---|---|---|---|
| No. | R1 | R2 | No. | drb | Yieldc (%) | |
| a All reactions were conducted using [Ph3C]+[B(C6F5)4]− (5.0 mol%) to generate 11, and silane (1.2 equiv.) and carbonyl compound (1.0 equiv.) with a ketone concentration of 0.2 M in CH2Cl2 at −60 °C. Reactions were terminated after 2½ h. b Diastereomeric ratio determined by GLC analysis prior to purification. c Combined yield of analytically pure mixture of diastereomers after flash chromatography. | ||||||
| 1 | 2j | –(CH2)4– | 13j | — | 85 | |
| 2 | 2k | –(CH2)5– | 13k | — | 95 | |
| 3 | 2l | –(CH2)11– | 13l | — | 86 | |
| 4 | 2m | Et | Et | 13m | — | 79 |
| 5 | 2n | Et | Me | 13n | 50∶50 |
44 |
| 6 | 2o | iBu | Me | 13o | 60∶40 |
79 |
| 7 | 2p | tBu | Me | 13p | 58∶42 |
77 |
These results are in contrast to the work of Kira et al.;8silylium ion-catalysed reduction of cyclododecanone (2l) afforded neither 13l nor cyclododecane but cyclododecene in high yield.11
To recap, we disclosed here an unprecedented silylium ion-catalysed reduction of carbonyl compounds, in which any of the previously reported deoxygenation pathways8,9 are suppressed. The hydride donor strength of our ferrocenyl-substituted silane is the decisive feature, allowing for the hydride transfer onto rather unreactive silylcarboxonium ions at low temperature. The reduction step itself regenerates the catalytically active trivalent silicon cation.
K.M. thanks the Studienstiftung des deutschen Volkes for a predoctoral fellowship (2009–2011). K.M. is a member of the International Research Training Group Münster–Nagoya (GRK 1143 of the Deutsche Forschungsgemeinschaft).
Notes and references
- For authoritative reviews, see: (a) J. B. Lambert, Y. Zhao and S. M. Zhang, J. Phys. Org. Chem., 2001, 14, 370–379 CrossRef CAS; (b) C. A. Reed, Acc. Chem. Res., 1998, 31, 325–332 CrossRef CAS; (c) for a recently developed silylium ion, see: S. Duttwyler, Q.-Q. Do, A. Linden, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2008, 47, 1719–1722 Search PubMed.
- H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39 10.1039/c003097j (Advance Article on April 19, 2010) and references cited therein.
- J. B. Lambert, Y. Zhao and H. Wu, J. Org. Chem., 1999, 64, 2729–2736 CrossRef CAS.
- (a) V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS; (b) R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676–9682 CrossRef CAS; (c) C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946–4953 CrossRef CAS.
- H. Mayr, N. Basso and G. Hagen, J. Am. Chem. Soc., 1992, 114, 3060–3066 CrossRef CAS.
- Mukaiyama-type processes, where a trivalent silicon cation is transferred between Lewis basic oxygen sites, are borderline cases. (a) [Et3Si(toluene)]+[B(C6F5)4]–-initiated Mukaiyama aldol reaction: K. Hara, R. Akiyama and M. Sawamura, Org. Lett., 2005, 7, 5621–5623 Search PubMed; (b) [Ph3C]+[B(C6F5)4]–-initiated polymerisation: Y. Zhang and E. Y.-X. Chen, Macromolecules, 2008, 41, 36–42 Search PubMed.
- For recent reviews of carbonyl hydrosilylation, see: (a) S. Rendler and M. Oestreich, in Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley, Weinheim, 2008, pp. 183–207 Search PubMed; (b) G. L. Larson and J. L. Fry, in Organic Reactions, ed. S. E. Denmark, Wiley, Hoboken, New Jersey, 2008, pp. 1–735 Search PubMed.
- M. Kira, T. Hino and H. Sakurai, Chem. Lett., 1992, 555–558 CrossRef CAS.
- D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS.
- Heteroatom-stabilisation of carbenium ions as in silylcarboxonium ions is expected to be significant. We sincerely thank Professor Ernst-Ulrich Würthwein for a preliminary quantum-chemical analysis of their hydride affinities.
- It is important to note that Kira et al. observed alkene instead of alkane formation with cyclododecanone, indicating a β-elimination of the intermediate oxonium ion (cf.6 in Scheme 1).8.
- (a) H. F. T. Klare, K. Bergander and M. Oestreich, Angew. Chem., Int. Ed., 2009, 48, 9077–9079 CrossRef CAS; (b) S. C. Bourke, M. J. MacLachlan, A. J. Lough and I. Manners, Chem.–Eur. J., 2005, 11, 1989–2000 CrossRef CAS.
- The drawing of 11 is likely to be an oversimplified representation of the true bonding situation. We are currently working on the structural characterisation of 11, supported by quantum-chemical calculations (in collaboration with S. Grimme and C. Mück-Lichtenfeld). For a related investigation in boron chemistry, see: M. Scheibitz, M. Bolte, J. W. Bats, H.-W. Lerner, I. Nowik, R. H. Herber, A. Krapp, M. Lein, M. C. Holthausen and M. Wagner, Chem.–Eur. J., 2005, 11, 584–603 Search PubMed.
- P. D. Bartlett, F. E. Condon and A. Schneider, J. Am. Chem. Soc., 1944, 66, 1531–1539 CrossRef CAS.
- Stereoinduction in the borohydride reduction of silicon-stereogenic silylcarboxonium ions was also found to be low: D. T. Hog and M. Oestreich, Eur. J. Org. Chem., 2009, 5047–5056 Search PubMed.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Preparation and characterisation data as well as 1H, 13C, 19F and 29Si NMR spectra of all new compounds. See DOI: 10.1039/c0cc02139c |
| § General procedure for the silylium ion-catalysed carbonylreduction: in a glove-box, a flame-dried 10 mL Schlenk tube equipped with a magnetic stir bar is charged with [Ph3C]+[B(C6F5)4]− (9.2 mg, 0.010 mmol, 5.0 mol%). The Schlenk tube is transferred to a fume cupboard and connected to an argon–vacuum manifold. Addition of dry CH2Cl2 (1.0 mL) results in a yellow solution, which is subsequently cooled to −60 °C using an ethanol cooling bath and a cryostat. After silane addition (10, 11 mg, 0.040 mmol, 0.20 equiv.), the now brown solution is stirred for 10 min, followed by successive addition of the carbonyl compound 2 (0.20 mmol, 1.0 equiv.) and the silane 10 (57 mg, 0.20 mmol, 1.0 equiv.) dissolved in dry CH2Cl2 (0.5 mL each). The reaction mixture is maintained at −60 °C for 2½ h, and the reaction is then terminated by the addition of dry hexane (10 mL), pre-cooled to −60 °C. Filtration over a small pad of Celite® and evaporation of the solvents under reduced pressure affords crude 13, which is further purified by flash chromatography on silica gel using cyclohexane as eluent. The diastereomeric ratio is determined by GLC analysis prior to purification. |
| This journal is © The Royal Society of Chemistry 2011 |