Detecting protein interactions in live cellsvia complementation of a hydrolysis-deficient β-lactamase†‡
Jiangsong
Jiang
a,
Junxiang
Zhang
b and
Shuwei
Li
*bc
aDepartment of Cell Biology and Molecular Genetics, University of Maryland, College Park, MD 20742, USA
bInstitute of Bioscience and Biotechnology Research, University of Maryland, Rockville, MD 20850, USA. E-mail: sli@umd.edu
cDepartment of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742, USA
First published on 10th August 2010
Abstract
Protein–fragment complementation assay using a hydrolysis-deficient β-lactamase provides a robust tool for studying protein–protein interactions.
Protein–protein interactions play a critical role in virtually all biological processes, including cell growth, differentiation, and communication. To understand interactions among various proteins, therefore, is a central theme of modern biology research. Over the past decades, numerous methods, such as yeast two-hybrid,1protein-fragment complementation assay (PCA),2 and tandem affinity purification,3 have been developed in order to decipher intricate protein interaction networks.
Ideally, protein interactions should be determined in their local compartments and physiological conditions, to prevent misidentification of binding events that are biologically irrelevant. PCA, in which nonfunctional N- and C-terminal fragments of a reporter protein fused with two interacting proteins can reconstitute its activity to generate a readout signal for detection, is one of such approaches that can probe protein interactions in their native environments. A variety of PCA techniques, each of which differs in the reporter protein and detection method,2,4 are currently available, offering a versatile set of choices for broad applications. For example, a green fluorescent protein (GFP) reporter can provide an optical signal for detecting protein interactions by fluorescent microscopy or fluorescent-activated cell sorting (FACS),5 while a dihydrofolate reductase (DHFR) based PCA allows binding proteins to be recognized through a cell-survival selection.6 Nevertheless, although PCA is a convenient tool for identifying unknown interacting proteins from a large library in bacteria7 or yeast,8 performing the same task in mammalian cells remains technically challenging as it usually requires virus transfection of cDNA libraries9 and screening strategies with single cell resolution.10
Herein, we describe a novel PCA based on a hydrolysis-deficient β-lactamase mutant (Lac-E166N),11 in hope of providing a simple protocol for the identification of associating proteins in live cells (Fig. 1). Wild type β-lactamase is a serine hydrolase that confers penicillin resistance in bacteria. It has already been widely used as a PCA reporter, which demonstrates many advantages, including small size (∼30 kD), no homologs in mammalian cells, high enzymatic activity, and availability of cell permeable substrates for fluorescent detection.12 This enzyme catalyzes the degradation of its substrates, β-lactam compounds, through a two-step mechanism. First, a β-lactam structure forms a stable covalent bond with Ser70. A water molecule activated by Glu166 then attacks this enzyme-substrate intermediate to turn over the enzyme. A point mutation at this position, such as E166N or E166A, impairs β-lactamase activity so that the enzyme will be trapped at acyl-enzyme intermediate due to its inability to remove the substrate from Ser70.13 Consequentially, reconstituted Lac-E166N can leave a permanent marker on the N-terminal fragment of β-lactamase when treated with a β-lactam substrate, even after both fragments dissociate. If a substrate containing an affinity tag is employed, the N-terminal fragment along with its fusion protein can be enriched by affinity purification and identified directly by mass spectrometry (MS), while a fluorescent substrate can make it compatible with fluorescent imaging and FACS screening.
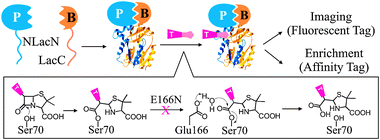 | ||
| Fig. 1 PCA using Lac-E166N. The catalytic mechanism of wild type β-lactamase and its hydrolysis-deficient mutant E166N is illustrated. P-prey protein; B-bait protein; T-tag. | ||
To test whether Lac-E166N can be used as a reporter for PCA applications, we first carried out an in vitro experiment using a pair of known interacting proteins (Fig. 2a). The N-terminal fragment of β-lactamase (residues 23–196) carrying E166N mutant was fused with the SH3 domain from yeast protein Sho1p (residues 302–367) through a flexible linker (GGGGS)3 to construct NLacN-SH3.14 The C-terminal fragment of β-lactamase (residues 198–286) was linked with a SH3 domain binding peptide (QQIVNKPLPPLPVAGSS, Kd = 800 nM) to form LacC-PPLP. We then incubated these protein fragments, either alone or in pair, with a fluorescent penicillin derivative (Flu-AP), and analyzed the reaction mixtures by SDS-PAGE. NLacN-SH3 became fluorescent only in the presence of LacC-PPLP, suggesting NLacN-SH3 was covalently modified by Flu-AP at Ser70 and the split fragments of this catalytically impaired Lac-E166N could refold into a functional structure upon association.
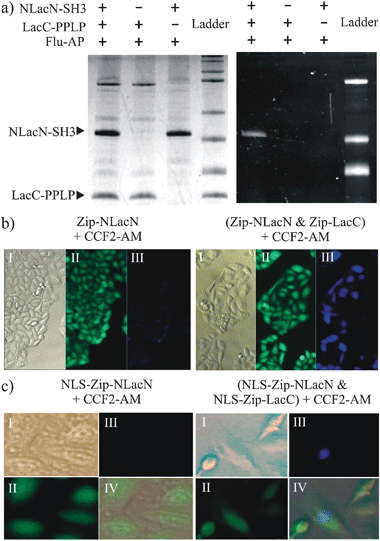 | ||
| Fig. 2 Lac-E166N as a PCA reporter in vitro and in vivo. (a) NLacN-SH3 and LacC-PPLP were incubated with Flu-AP either alone or in pair and analyzed by SDS-PAGE. Coomassie blue staining (left); Fluorescent imaging (right). (b) HeLa cells expressing Zip-NLacN alone (left) or both Zip-NLacN and Zip-LacC (right) were treated with CCF2-AM. (c) HeLa cells expressing NLS-Zip-NLacN alone (left) or both NLS-Zip-NLacN and NLS-Zip-LacC (right) were treated with CCF2-AM.The parentheses indicate both fragments were expressed by the same plasmid. I-DIC (differential interference contrast); II-green channel; III-blue channel; IV-overlay of I, II, and III. | ||
Next, we examined if Lac-E166N fragments can recover its function inside live cells. In order to compensate low transfection efficiency in mammalian cells, we constructed one plasmid that expresses both NLacN and LacC fragments fused to the C-terminus of a leucine zipper domain adopted from yeast GCN4 protein (Zip-NLacN and Zip-LacC). This leucine zipper domain can form a homodimer and is often used as a model system for testing protein interactions.15 After HeLa cells were transfected with this plasmid, a cell permeable β-lactamase substrate, CCF2-AM (AM = acetoxymethyl), was added to detect the activity of reconstituted Lac-E166N (Fig. 2b). CCF2-AM is a fluorescence resonance energy transfer (FRET) based molecule, which emits green light (518 nm) at its intact form, yet becomes blue fluorescent (447 nm) upon enzymatic degradation.16 We observed that about 60% HeLa cells transfected with this plasmid exhibited blue fluorescence. In contrast, none of cells displayed blue fluorescence when only Zip-NLacN fragment was expressed, indicating blue fluorescence resulted from the interaction between two β-lactamase fragments induced by dimerization of leucine zipper domain.
Because CCF2-AM leaves a fluorescent tag on the Ser70 of NLacN after reaction, this PCA strategy would also allow us to track the movement of a protein after it interacts with a partner. To confirm this, we inserted a nuclear localization signal before the leucine zipper domain so both fragments (NLS-Zip-NLacN and NLS-Zip-LacC) should travel to nuclei. When these fragments were expressed in HeLa cells, we found blue fluorescent NLS-Zip-NLacN was indeed concentrated in nuclei (Fig. 2c). At this moment, however, we could not determine whether NLS-Zip-NLacN interacted with NLS-Zip-LacC in nuclei or moved into nuclei after binding with NLS-Zip-LacC in cytoplasm.
Compared to wild type β-lactamase based PCA, one advantage of using hydrolysis-deficient Lac-E166N as a reporter is that proteins interacting with a bait could be identified with MS directly (Fig. 3a). To demonstrate this idea, we synthesized a novel cell permeable prodrug substrate of β-lactamase, CP-AM, which contains a terminal alkyne that can react with azide bioorthogonally. This compound was added into a HEK293T cell culture expressing Zip-NLacN and Zip-LacC. Once entering cells, CP-AM became active after its protective AM group was removed by nonspecific cellular esterases and formed a stable covalent bond with Ser70 in the Zip-NLacN. Cells were then collected, lysed, and treated with solid support beads coated with azide. In the presence of Cu(I) catalyst, labeled Zip-NLacN was immobilized covalently on beads, washed thoroughly, then digested by trypsin. The resulting peptides were analyzed by LC-MS/MS and Mascot for protein identification. We found peptides from both β-lactamase and leucine zipper domain (Fig. 3b and c) with minimum background from other endogenous proteins, suggesting this affinity enrichment protocol was highly efficient.
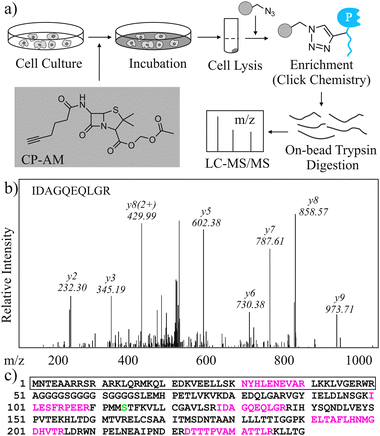 | ||
| Fig. 3 Discovery of protein interactions using MS in Lac-E166N based PCA. (a) Scheme of how interacting proteins are enriched and identified. (b) A representative peptide from enriched Zip-NLacN was identified by LC-MS/MS and Mascot. The sequence of this peptide and its b-/y- ions are marked. (c) Peptide sequence of Zip-NLacN. Tryptic peptides identified by LC-MS/MS and Mascot are coloured in red and Ser70 is coloured in green. The sequence in the box is from leucine zipper domain. | ||
By introducing a single point mutation into β-lactamase, we were able to establish a new PCA system that can be applied on either fluorescent imagining or affinity enrichment of interacting proteins by using different substrates. Although this method is not as sensitive as wild type β-lactamase based PCA since the readout signal is not enzymatically amplified, it would allow protein interactions to be detected at their local sites, providing valuable information on their biological functions. In addition, this approach would potentially enable us to discover unknown binding proteins from a large cDNA library. Finally, because protein interacting events can leave a permeant tag on the NLacN fragment even after protein complexes dissociate, it would be of great interest to explore whether this technique can detect transient and low-affinity protein–protein interactions.
Notes and references
- J. R. Parrish, K. D. Gulyas and R. L. Finley, Jr., Curr. Opin. Biotechnol., 2006, 17, 387–393 CrossRef CAS; S. Fields and O.-k. Song, Nature, 1989, 340, 245–246 CrossRef CAS.
- M. Morell, S. Ventura and F. X. Aviles, FEBS Lett., 2009, 583, 1684–1691 CrossRef CAS.
- T. Burckstummer, K. L. Bennett, A. Preradovic, G. Schutze, O. Hantschel, G. Superti-Furga and A. Bauch, Nat. Methods, 2006, 3, 1013–1019 CrossRef.
- I. Remy and S. W. Michnick, Nat. Methods, 2006, 3, 977–979 CrossRef CAS; P. A. Vidi and V. J. Watts, Mol. Pharmacol., 2009, 75, 733–739 CrossRef CAS.
- I. Remy and S. W. Michnick, Methods Enzymol., 2004, 32, 381–388 CrossRef CAS.
- S. Shibasaki, K. Sakata, J. Ishii, A. Kondo and M. Ueda, Appl. Microbiol. Biotechnol., 2008, 80, 735–743 CrossRef CAS.
- J. H. Park, J. H. Back, S. H. Hahm, H. Y. Shim, M. J. Park, S. I. Ko and Y. S. Han, J. Microbiol. Biotechnol., 2007, 17, 1607–1615 CAS.
- K. Tarassov, V. Messier, C. R. Landry, S. Radinovic, M. M. Molina, I. Shames, Y. Malitskaya, J. Vogel, H. Bussey and S. W. Michnick, Science, 2008, 320, 1465–1470 CrossRef CAS; U. Stelzl, U. Worm, M. Lalowski, C. Haenig, F. H. Brembeck, H. Goehler, M. Stroedicke, M. Zenkner, A. Schoenherr, S. Koeppen, J. Timm, S. Mintzlaff, C. Abraham, N. Bock, S. Kietzmann, A. Goedde, E. Toksoz, A. Droege, S. Krobitsch, B. Korn, W. Birchmeier, H. Lehrach and E. E. Wanker, Cell, 2005, 122, 957–968 CrossRef CAS; J. F. Rual, K. Venkatesan, T. Hao, T. Hirozane-Kishikawa, A. Dricot, N. Li, G. F. Berriz, F. D. Gibbons, M. Dreze, N. Ayivi-Guedehoussou, N. Klitgord, C. Simon, M. Boxem, S. Milstein, J. Rosenberg, D. S. Goldberg, L. V. Zhang, S. L. Wong, G. Franklin, S. Li, J. S. Albala, J. Lim, C. Fraughton, E. Llamosas, S. Cevik, C. Bex, P. Lamesch, R. S. Sikorski, J. Vandenhaute, H. Y. Zoghbi, A. Smolyar, S. Bosak, R. Sequerra, L. Doucette-Stamm, M. E. Cusick, D. E. Hill, F. P. Roth and M. Vidal, Nature, 2005, 437, 1173–1178 CrossRef CAS.
- Z. Ding, J. Liang, Y. Lu, Q. Yu, Z. Songyang, S. Y. Lin and G. B. Mills, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15014–15019 CrossRef CAS.
- E. Kiss-Toth, F. M. Guesdon, D. H. Wyllie, E. E. Qwarnstrom and S. K. Dower, J. Immunol. Methods, 2000, 239, 125–135 CrossRef CAS.
- G. Minasov, X. Wang and B. K. Shoichet, J. Am. Chem. Soc., 2002, 124, 5333–5340 CrossRef CAS.
- T. Wehrman, B. Kleaveland, J. H. Her, R. F. Balint and H. M. Blau, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3469–3474 CrossRef CAS; A. Galarneau, M. Primeau, L. E. Trudeau and S. W. Michnick, Nat. Biotechnol., 2002, 20, 619–622 CrossRef CAS.
- H. Adachi, T. Ohta and H. Matsuzawa, J. Biol. Chem., 1991, 266, 3186–3191 CAS.
- J. A. Marles, S. Dahesh, J. Haynes, B. J. Andrews and A. R. Davidson, Mol. Cell, 2004, 14, 813–823 CrossRef CAS.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, Science, 1993, 262, 1401–1407 CAS.
- G. Zlokarnik, P. A. Negulescu, T. E. Knapp, L. Mere, N. Burres, L. Feng, M. Whitney, K. Roemer and R. Y. Tsien, Science, 1998, 279, 84–88 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experiment procedures. See DOI: 10.1039/c0cc01998d |
| This journal is © The Royal Society of Chemistry 2011 |