Singlet oxygen initiated cascade transformation of a simple difuran into the key ABC-ring motif of the pectenotoxins†‡
Georgios
Vassilikogiannakis
*,
Ioanna
Alexopoulou
,
Maria
Tofi
and
Tamsyn
Montagnon
Department of Chemistry, University of Crete, 71003 Iraklion, Crete, Greece. E-mail: vasil@chemistry.uoc.gr; Fax: +30 2810 545001; Tel: +30 2810 545074
First published on 26th July 2010
Abstract
The key ABC-ring motif of the pectenotoxins has been synthesized, starting from a simple and readily accessible difuran precursor, using a complex singlet oxygen-mediated cascade reaction sequence.
As a continuation of our interest in the development of new and efficient synthetic approaches to natural products1 and important natural products motifs2 based on implementing cascade reaction sequences including, as the key element, singlet oxygen-mediated photooxygenations,3 we sought to explore the possibility of synthesising the key ABC-ring system of polyether macrolactones pectenotoxins 2c, 8, 9 and 11c (Fig. 1) using such chemistry.
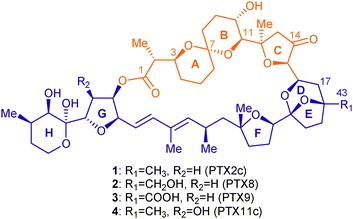 | ||
| Fig. 1 Structures of PTXs 2c, 8, 9 and 11c. | ||
Since the first isolation of members of the pectenotoxin (PTX) family in 1985 from Patinopecten yessoensis scallops, by Yasumoto and his coworkers,4 more than 20 other structurally related compounds have been isolated from Dinophysis dinoflagellates worldwide.5 PTXs have aroused particular interest because they interact with the actin cytoskeleton at a new and unique site6 conferring on them potent cytotoxicity against human lung, colon and breast cancer cell lines.5a,7 Excluding subtle variations in the oxidation state at C43 (Fig. 1), PTXs structural diversity originates mainly from differing types and configurations of AB spiroketal subunit that are interconvertible by means of acid equilibration.5b Only four PTXs bear a [6,6]-spiroketal, while in the rest a [6,5]-spiroketal is present.
PTX4 and PTX8 were synthesised in 2002 with characteristic eloquence by Evans and coworkers.8 The ABC-fragment, containing a [6,5]-spiroketal, has been synthesised by the groups of Paquette,9 Brimble10 and Pihko11 using acid catalysed ketalisation of hydroxyketones, or ketals, for the formation of the C7 spiroketal, as well as a classical 5-exo epoxide opening for the construction of the C-ring.12 More exotic approaches, namely the cyclization of a spirodiepoxide and reductive cyclization of a cyanoacetal for the synthesis of the [6,5] AB-spiroketal, have recently been developed by the groups of Williams13 and Rychnovsky,14 respectively.
The main focus of work emanating from our laboratories has been to explore new ways of employing singlet oxygen, a powerful, selective and green oxidant, as a cascade reaction sequence-mediator.1–3 To this end, we have recently reported methods to rapidly synthesise [6,5]- and [6,6]-spiroketals,2c,d as well as to make 3-keto-tetrahydrofurans2e using singlet oxygen (1O2) oxidation of an appropriately substituted simple furan precursor. Based on this experience gained designing and implementing these cascade reaction sequences, we thought it might be possible, although definitely ambitious, to combine the methodologies in one super-cascade sequence mediated by 1O2 that would yield the key ABC-ring motif (5, Scheme 1) of PTXs 2c, 8, 9 and 11c starting from a relatively simple difuran precursor 6.
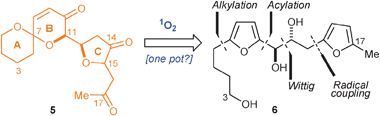 | ||
| Scheme 1 Retrosynthetic analysis of ABC motif of PTXs. | ||
Our investigation began by targeting the cascade reaction sequence precursor, difuran-triol 6. We hoped to achieve the synthesis of 6 using syn-dihydroxylation of a cis-olefin, to be prepared by means of a Wittig reaction (Scheme 1). According to precedent,2c,15 we believed it should be possible to introduce all the requisite substituents at the ortho positions of the difuran 6 using known anionic- or radical-based methods at an early stage in the synthetic sequence.
The strategy was implemented, and, in a straightforward manner, free radical coupling of an excess of commercially available sylvan (7) with ethyl iodoacetate according to Baciocchi conditions15 afforded ethyl 2-(furan-2-yl)acetate (8, Scheme 2) in 91% yield. Reduction of 815b with LiAlH4 followed by iodination (I2, PPh3, imidazole) gave iodide 9, which was converted to the corresponding phosphonium salt 10 in 68% overall yield (over 3 steps). Wittig coupling between the phosphonium salt 10 and aldehyde 13 (prepared by ortho-formylation of known furan 122c,d with DMF) afforded olefin 11, as a mixture of two geometrical isomers (cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans = 2:1, Scheme 2), in 73% yield. Sharpless asymmetric dihydroxylation (SAD)16 of olefin 11 (cis:trans = 2:1) resulted in the unexpected formation of a 1:2.6 mixture of erythro/threo 1,2-diols17 in 62% yield, while 20% of the starting material (exclusively cis-11) was recovered. The unexpected formation of the threo diastereoisomer of 6 as the major product of this syn-dihydroxylation reaction of an olefin whose major geometrical isomer is cis configured, implies a degree of cis/transisomerisation during the course of the reaction. The fact that the recovered starting olefin is exclusively cis is in agreement with the known faster dihydroxylation of a transolefinversus its cis isomer. When the recovered cis-11 was resubjected to SAD conditions a 2.5:1 mixture of erythro/threo 1,2-diols was formed. Finally, TBAF assisted deprotection of the primary alcohol resulted in the formation of the requisite photooxygenation precursor difuran-triol 6 in a total of eight simple synthetic steps.
:trans = 2:1, Scheme 2), in 73% yield. Sharpless asymmetric dihydroxylation (SAD)16 of olefin 11 (cis:trans = 2:1) resulted in the unexpected formation of a 1:2.6 mixture of erythro/threo 1,2-diols17 in 62% yield, while 20% of the starting material (exclusively cis-11) was recovered. The unexpected formation of the threo diastereoisomer of 6 as the major product of this syn-dihydroxylation reaction of an olefin whose major geometrical isomer is cis configured, implies a degree of cis/transisomerisation during the course of the reaction. The fact that the recovered starting olefin is exclusively cis is in agreement with the known faster dihydroxylation of a transolefinversus its cis isomer. When the recovered cis-11 was resubjected to SAD conditions a 2.5:1 mixture of erythro/threo 1,2-diols was formed. Finally, TBAF assisted deprotection of the primary alcohol resulted in the formation of the requisite photooxygenation precursor difuran-triol 6 in a total of eight simple synthetic steps.
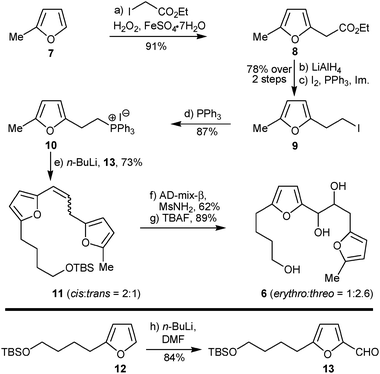 | ||
| Scheme 2 Synthesis of the photooxygenation precursor difuran-triol 6. | ||
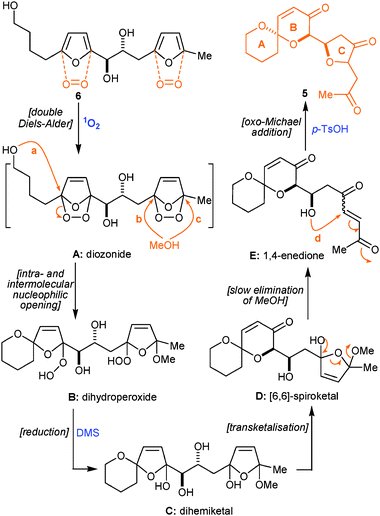
With difuran-triol 6 in hand, the stage was now set for us to obtain proof of principle for the ambitious singlet oxygen-initiated cascade reaction that we had envisioned as a means of accessing the key ABC-ring motif of the pectenotoxins. A solution of 6 (erythro/threo = 1:2.6) in MeOH, containing rose bengal as photosensitizer, was irradiated for three minutes with visible spectrum light whilst oxygen was being bubbled gently through it. Very careful replacement of MeOH with either CHCl3 or CH2Cl2 followed by treatment for 72 h with an excess of dimethyl sulfide (DMS) and then with p-TsOH for 3 h resulted in the remarkable formation of compound 14 (51%, mixture of 3 diastereoisomers in 5:3:2 ratio, Scheme 3) accompanied by a chromatographically separable, less polar, compound 5 (18%, mixture of 2 diastereoisomers in 7:3 ratio). When the same photooxidation conditions were applied to 6 (erythro/threo = 2.5:1) formation of compound 5 (49%, mixture of 2 diastereoisomers in 7:3 ratio) was accompanied by compound 14 (19%, mixture of 3 diastereoisomers in 5:3:2 ratio). Careful NOE studies undertaken on the major diastereoisomer of 14 (observation of NOE between H3 and H11, as well as H12 and H15) prove that the relative stereochemistries of C7 compared to C11, as well as C12 compared to C15 are correct. Similar NOE studies on 5 (observation of NOE between H3 and H11) are consistent with the anomeric C7 spiroketal for both diastereoisomers.18
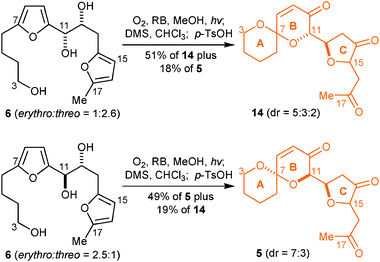 | ||
| Scheme 3 Singlet oxygen initiated cascade transformation of difuran-triol 6 into the ABC motif of PTXs 2c, 8, 9, and 11c. | ||
The synthetic strategy for PTXs ABC-ring motif synthesis that had just been successfully implemented, had been predicated on our previous experience in the field1a–c,g,2c–e which would also support the following mechanistic rationale (Scheme 4) for the transformation of 6 into 5. First of all, the formation of dihydroperoxide B (Scheme 4) is expected to occur via one intra- and one intermolecular nucleophilic opening of fleeting diozonide A. For simplicity, only one of the two possible regioisomers (nucleophilic openings b and c) of dihydroperoxide B appears in Scheme 4. Each regioisomer is in turn a mixture of diastereoisomers resulting in a very complicated 1H NMR spectrum for dihydroperoxide B. The very careful replacement of MeOH with CHCl3 followed by treatment of the mixture of dihydroperoxides B with an excess of dimethyl sulfide (DMS) reduces1a–c,2c–e the two hydroperoxy groups of B, thus, affording the corresponding dihemiketal C. A transketalisation reaction2d leads to the transformation of the [6,5]-spiroketal of C into the [6,6]-spiroketal D. Under the same reaction conditions the remaining hemiketal moiety of intermediate D slowly eliminates a molecule of MeOH,1a–c,2e thus revealing the 1,4-enedione moiety of intermediate E. In the final step of this remarkable cascade sequence, p-TsOH catalyses an oxo-Michael cyclisation2e between the –OH group of E and the 1,4-enedione moiety resulting in the formation of the sought after ABC-ring motif of PTXs in the form of compound 5.
 | ||
| Scheme 4 A mechanistic explanation. | ||
From a practical standpoint, it is very important to note that during the 72 h treatment of the photooxidation mixture with DMS the reaction was continuously monitored by 1H NMR. The fact that the intermediates B, C, D and E (Scheme 4) are mixtures of different regio- (not shown) and stereoisomers, and also the fact that some of these intermediates coexist during the course of the reaction, made analysis of the 1H NMR spectra challenging. However, it is very important to undertake this monitoring by 1H NMR because not only does the DMS/DMSO ratio remain unchanged once complete reduction of all the hydroperoxy groups has been attained, but the amount of MeOH present also becomes static once the opening of the hemiketal to the 1,4-enedione moiety has been achieved, thus the right moment for the addition of the p-TsOH is indicated. This final reagent addition initiates a gross simplification of the reaction's TLC profile that has been complex up to this point, as two less polar (compared to preceding intermediates) spots start to dominate.
In summary, we have designed and successfully executed a most ambitious singlet oxygen-driven reaction cascade sequence wherein a simple and readily accessible difuran precursor is transformed to a complex multiringed motif found in the pectenotoxin family of natural products. This sequence perfectly showcases the power singlet oxygen has to mediate intricate cascade reaction sequences and transform simple molecules to complex ones with ease and efficiency.
This research was supported by a Marie Curie European Integration Grant (T.M.), within the 7th European Community Framework Programme, ELKE of the University of Crete (K.A. 2949), as well as COST action CM0804.
Notes and references
- (a) G. Vassilikogiannakis and M. Stratakis, Angew. Chem., Int. Ed., 2003, 42, 5465–5468 CrossRef CAS; (b) G. Vassilikogiannakis, I. Margaros and T. Montagnon, Org. Lett., 2004, 6, 2039–2042 CrossRef CAS; (c) G. Vassilikogiannakis, I. Margaros, T. Montagnon and M. Stratakis, Chem.–Eur. J., 2005, 11, 5899–5907 CrossRef CAS; (d) I. Margaros and G. Vassilikogiannakis, J. Org. Chem., 2007, 72, 4826–4831 CrossRef; (e) I. Margaros, T. Montagnon and G. Vassilikogiannakis, Org. Lett., 2007, 9, 5585–5588 CrossRef CAS; (f) I. Margaros and G. Vassilikogiannakis, J. Org. Chem., 2008, 73, 2021–2023 CrossRef CAS; (g) E. Pavlakos, T. Georgiou, M. Tofi, T. Montagnon and G. Vassilikogiannakis, Org. Lett., 2009, 11, 4556–4559 CrossRef CAS.
- (a) N. Sofikiti, M. Tofi, T. Montagnon, G. Vassilikogiannakis and M. Stratakis, Org. Lett., 2005, 7, 2357–2359 CrossRef CAS; (b) I. Margaros, T. Montagnon, M. Tofi, E. Pavlakos and G. Vassilikogiannakis, Tetrahedron, 2006, 62, 5308–5317 CrossRef; (c) T. Georgiou, M. Tofi, T. Montagnon and G. Vassilikogiannakis, Org. Lett., 2006, 8, 1945–1948 CrossRef CAS; (d) M. Tofi, T. Montagnon, T. Georgiou and G. Vassilikogiannakis, Org. Biomol. Chem., 2007, 5, 772–777 RSC; (e) M. Tofi, K. Koltsida and G. Vassilikogiannakis, Org. Lett., 2009, 11, 313–316 CrossRef CAS.
- For reviews on furan photooxygenation see: (a) K. Gollnick and A. Griesbeck, Tetrahedron, 1985, 41, 2057–2068 CrossRef CAS; (b) B. L. Feringa, Recl. Trav. Chim. Pays-Bas, 1987, 106, 469–488 CAS; (c) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001–1011 CrossRef CAS.
- T. Yasumoto, M. Murata, Y. Oshima, M. Sano, G. K. Matsumoto and J. Clardy, Tetrahedron, 1985, 41, 1019–1025 CrossRef CAS.
- For selected examples see: (a) J. H. Jung, C. J. Sim and C. O. Lee, J. Nat. Prod., 1995, 58, 1722–1726 CrossRef CAS; (b) K. Sasaki, J. L. C. Wright and T. Yasumoto, J. Org. Chem., 1998, 63, 2475–2480 CrossRef CAS; (c) M. Daiguji, M. Satake, K. James, A. Bishop, L. Mackenzie, H. Naoki and T. Yasumoto, Chem. Lett., 1998, 653–654 CrossRef CAS; (d) A. L. Wilkins, N. Rehmann, T. R. Torgersen, M. Keogh, D. Petersen, P. Hess, F. Rise and C. O. Miles, J. Agric. Food Chem., 2006, 54, 5672–5678 CrossRef CAS; (e) C. O. Miles, A. L. Wilkins, A. D. Hawkes, D. J. Jensen, A. I. Selwood, V. Beuzenberg, A. L. MacKenzie, J. M. Cooney and P. T. Holland, Toxicon, 2006, 48, 152–159 CrossRef CAS; (f) P. Kuuppo, P. Uronen, A. Petermann, T. Tamminen and E. Granéli, Limnol. Oceanogr., 2006, 51, 2300–2307 CrossRef CAS.
- (a) I. Spector, F. Braet, N. R. Schochet and M. R. Bubb, Microsc. Res. Tech., 1999, 47, 18–37 CrossRef CAS; (b) F. Leira, A. G. Cabadon, M. R. Vieytes, Y. Roman, A. Alfonso, L. M. Botana, T. Yasumoto, C. Malaguti and G. P. Rossini, Biochem. Pharmacol., 2002, 63, 1979–1988 CrossRef CAS.
- V. Burgess and G. Shaw, Environ. Int., 2001, 27, 275–283 CrossRef CAS.
- (a) D. A. Evans, H. A. Rajapakse and D. Stenkamp, Angew. Chem., Int. Ed., 2002, 41, 4569–4573 CrossRef CAS; (b) D. A. Evans, H. A. Rajapakse, H. A. Chiu and D. Stenkamp, Angew. Chem., Int. Ed., 2002, 41, 4573–4576 CrossRef CAS.
- (a) D. Bondar, J. Liu, T. Muller and L. A. Paquette, Org. Lett., 2005, 7, 1813–1816 CrossRef CAS; (b) P. D. O’Connor, C. K. Knight, D. Friedrich, X. Peng and L. A. Paquette, J. Org. Chem., 2007, 72, 1747–1754 CrossRef CAS.
- (a) R. Halim, M. A. Brimble and J. Merten, Org. Lett., 2005, 7, 2659–2662 CrossRef CAS; (b) R. Halim, M. A. Brimble and J. Merten, Org. Biomol. Chem., 2006, 4, 1387–1399 RSC.
- (a) P. M. Pihko and J. E. Aho, Org. Lett., 2004, 6, 3849–3852 CrossRef CAS; (b) J. E. Aho, E. Salomäki, K. Rissanen and P. M. Pihko, Org. Lett., 2008, 10, 4179–4182 CrossRef CAS; (c) H. Helmboldt, J. E. Aho and P. M. Pihko, Org. Lett., 2008, 10, 4183–4185 CrossRef CAS.
- For a review about all the synthetic studies towards PTXs until 2006, see: R. Halim and M. A. Brimble, Org. Biomol. Chem., 2006, 4, 4048–4058 Search PubMed.
- S. D. Lotesta, Y. Hou and L. J. Williams, Org. Lett., 2007, 9, 869–872 CrossRef CAS.
- (a) D. Vellucci and S. D. Rychnovsky, Org. Lett., 2007, 9, 711–714 CrossRef CAS; (b) L. R. Takaoka, A. J. Buckmelter, T. E. La Cruz and S. D. Rychnovsky, J. Am. Chem. Soc., 2005, 127, 528–529 CrossRef CAS.
- (a) E. Baciocchi, E. Muraglia and G. Sleiter, J. Org. Chem., 1992, 57, 6817–6820 CrossRef CAS; (b) F. Loiseau, J.-M. Simone, D. Carcache, P. Bobal and R. Neier, Monatsh. Chem., 2007, 138, 121–129 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- For the threo, erythro assignment of similar compounds see: (a) W. Adam and B. Nestler, J. Am. Chem. Soc., 1993, 115, 5041–5049 CrossRef CAS; (b) G. Vassilikogiannakis, M. Stratakis, M. Orfanopoulos and C. S. Foote, J. Org. Chem., 1999, 64, 4130–4139 CrossRef CAS.
- For detailed NOE studies see supporting information section.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c0cc01341b |
| This journal is © The Royal Society of Chemistry 2011 |