Luminescent probes based on water-soluble, dual-emissive lanthanide complexes: metal ion-induced modulation of near-IR emission†‡
Michael
Andrews
a,
Jennifer E.
Jones
a,
Lindsay P.
Harding
b and
Simon J. A.
Pope
*a
aSchool of Chemistry, Main Building, Cardiff University, Cardiff, Cymru/Wales, UK CF10 3AT. E-mail: popesj@cardiff.ac.uk; Fax: +44 (0)2920874030; Tel: +44 (0)2920879316
bDepartment of Biological and Chemical Sciences, University of Huddersfield, Queensgate, Huddersfield, UK HD1 3DH
First published on 15th June 2010
Abstract
Responsive lanthanide complexes demonstrate differentiated modulated luminescence output upon exposure to metal di-cations in aqueous solution.
Due to their unique and advantageous physical properties, lanthanide ion complexes offer numerous opportunities when considering the design of responsive molecular probes.1 Their successful exploitation within biological imaging modalities, principally as luminophores2 and magnetic resonance (MR) contrast agents,3 implies that continued effort is required to maximize their potential in this regard: biologically compatible imaging agents that can report upon their environment will be a crucial non-invasive tool in neurobiology.4 An approach to rationally design responsive (lanthanide-based) probes, mutually addressing luminescence and MR modalities, should consider the inner coordination sphere of the lanthanide (specifically the hydration state, q). The vibrational manifold of O–H oscillators provides a very efficient non-radiative quenching pathway for f-centred excited states, whilst the longitudinal proton relaxivity of aqueous GdIII complexes is dictated, in part, by the accessibility of water to the GdIII centre.
In this communication we focus upon the design, syntheses and luminescent properties of hetero-chromophoric lanthanide complexes. The complexes incorporate secondary binding sites for targeting metal di-cations of physiological and toxicological importance: CuII, ZnII, CdII and HgII. The binding site also hosts a long-wavelength chromophore enabling: (i) the potential for dual emission; (ii) the visible sensitization of near-IR emitting lanthanide ions such as NdIII and YbIII;5 and (iii) the potential for single molecule bimodal fluorescent/MR agents.
The synthetic strategy required the preformation of the chromophoric moieties, thus 8-aminoquinoline and 1-amino-9,10-anthraquinone both reacted with chloroacetyl chloride to give the corresponding chloroacetamides. Stoichiometric (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1) coupling with picolylamine gave the corresponding chromophore-appended secondary amines (a–b), which were then alkylated using the chloromethylazamacrocyclic derivative 1 (Scheme 1).6 Our studies have shown that the use of 1 allows bulky architectures to be efficiently coupled: the alternative strategy of alkylating 1,4,7-tris(tert-butoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecane7 required long reaction times and gave poor yields. The amide linkage proved sufficiently robust to subsequent deprotection strategies, preventing cleavage of the aryl units. Deprotection (1∶1, trifluoroacetic acid/CH2Cl2) yielded the free chromophore-appended ligands as highly hygroscopic TFA adducts (quantitatively determined by elemental analyses)8La–ba–b and subsequent complexation with Ln(OTf)3 gave the corresponding lanthanide complexes, Ln–La–ba–b (Ln = NdIII, EuIII, GdIII, and YbIII). Full experimental details and comprehensive spectroscopic characterisation for all ligand precursors and intermediates, deprotected ligands and lanthanide complexes are provided within the ESI.†
∶1) coupling with picolylamine gave the corresponding chromophore-appended secondary amines (a–b), which were then alkylated using the chloromethylazamacrocyclic derivative 1 (Scheme 1).6 Our studies have shown that the use of 1 allows bulky architectures to be efficiently coupled: the alternative strategy of alkylating 1,4,7-tris(tert-butoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecane7 required long reaction times and gave poor yields. The amide linkage proved sufficiently robust to subsequent deprotection strategies, preventing cleavage of the aryl units. Deprotection (1∶1, trifluoroacetic acid/CH2Cl2) yielded the free chromophore-appended ligands as highly hygroscopic TFA adducts (quantitatively determined by elemental analyses)8La–ba–b and subsequent complexation with Ln(OTf)3 gave the corresponding lanthanide complexes, Ln–La–ba–b (Ln = NdIII, EuIII, GdIII, and YbIII). Full experimental details and comprehensive spectroscopic characterisation for all ligand precursors and intermediates, deprotected ligands and lanthanide complexes are provided within the ESI.†
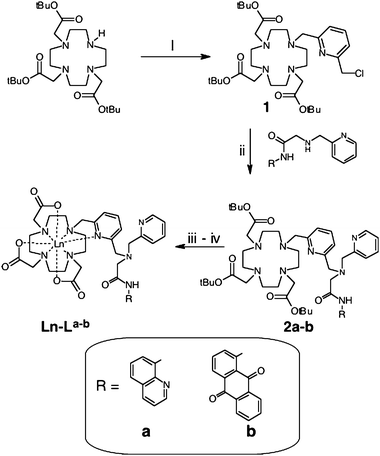 | ||
| Scheme 1 Synthetic pathway to the ligands and complexes. Reagents and conditions: (i) 1.1 eq. 2,6-bis(chloromethyl)pyridine, MeCN, NaHCO3, Δ; (ii) 1 eq. a or b, MeCN, NaHCO3, KI, Δ; (iii) CF3CO2H, CH2Cl2; and (iv) Ln(OTf)3, MeOH, Δ. | ||
The UV-vis absorption properties of the free ligands are dominated by the superimposition of bands originating from the aromatic units. As anticipated the anthraquinone derivative, Lbb, demonstrated absorption well into the visible region, with a broadened intraligand-centred band ca. 400 nm (ε > 103 M−1 cm−1) attributed to significant charge transfer character (formally N-to-quinone).9 Ligand-centred features also dominate the absorption spectra of the corresponding complexes.
Both ligands were fluorescent in solution with emission profiles (λex = 355 nm) and wavelengths characteristic of the fluorophores. Room temperature emission spectra showed the expected ligand-centred fluorescence for the complexes (ESI†, Fig. S1), with Gd–Lb particularly noteworthy (λmax = 462 nm). Measurements on Gd–Laa at 77 K (2∶1 ethanol∶methanol glass) allowed the triplet state (ESI†, Fig. S1) of the amido-quinoline chromophore to be identified at ca. 20800 cm−1 (the amide-functionalised anthraquinone chromophore is ca. 22000 cm−1).9a Therefore both are of sufficient energy to populate the f-centred excited states of NdIII and YbIII, assuming sensitization predominantly occurs via ligand-centred triplet states.5 For EuIII (5D0 state ca. 17250 cm−1), sensitization by 1-amido-9,10-anthraquinone and 8-amido-quinoline also appears feasible on energetic grounds, although dramatically increased EuIII emission intensity was observed via pyridyl excitation, reflecting the spatial proximity of the latter.
The NdIII and YbIII complexes demonstrated dual emission (λex = 355 nm): visible, ligand-centred fluorescence together with near-IR lanthanide emission. The luminescence lifetimes were characteristic (in water and D2O) of lanthanide complexes of this ligand type. It is important to note that absorption at 355 nm is due to the long-wavelength chromophores within each ligand: the pyridyl units do not absorb at this wavelength, and therefore sensitization of YbIII and NdIII must occur via a through space energy transfer mechanism. The corresponding steady state spectra were also typical with the NdIII complexes displaying near-IR emission dominated by a peak at 1058 nm (4F3/2 → 4I11/2) with a weaker feature at ca. 1340 nm, corresponding to 4F3/2 → 4I13/2, whilst Yb–La–b gave profiles with vibronically structured peaks at ca. 980 nm (2F5/2 → 2F7/2). Complexes of Lbb demonstrate that visible-light excitation wavelengths up to 425 nm can be employed to sensitize lanthanide emission.9a
The single exponential luminescence lifetimes for the YbIII and NdIII complexes are collected in Table 1 and were utilised to deduce the inner sphere hydration, q, of each lanthanide ion [q = A{(kH2O – kD2O) – B}; for YbIIIA = 1 µs, B = 0.2 µs−1; for aqueous NdIIIq = 130 (kH2O – kD2O) − 0.4].10 The values show that water is effectively shielded from the lanthanide coordination sphere, suggesting an octadentate donor-set contribution from the ligands (i.e. the bridging pyridine is coordinated to the lanthanide ion),6,8a,11 supplemented with the inherent hydrophobicity of the chromophoric binding sites. Unfortunately the relatively poor emissivity of the sensitised complexes prevented reliable quantum yield measurements from being obtained. The overall quantum yield is related to the effectiveness of (visible) sensitisation and the intrinsic quantum yield (i.e. QLnL = ηsens × QLnLn),12 where the intrinsic quantum yield can be approximated by τobs/τrad (using literature τrad values in D2O for NdIII and YbIII of 270 µs and 1.21 ms respectively).13 For Nd/Yb–La–b the estimated QLnLn values in D2O are 0.1–0.6% implying that energy transfer from the long wavelength antennae is very inefficient, which is not unexpected for a remote, uncoordinated chromophore,14 thus reducing the overall quantum yield.
| Compound | τ ( H 2 O )/µs | τ ( D 2 O )/µs | q b |
|---|---|---|---|
| a Errors on lifetime measurements are approximately ±10%. b q = A{(kH2O – kD2O) – B}; for YbIIIA = 1 µs, B = 0.2 µs−1; for aqueous NdIIIq = 130 (kH2O – kD2O) − 0.4.10λex = 355 nm. | |||
| Nd–Laa | 0.144 | 0.430 | 0.6 |
| Nd–Lbb | 0.129 | 0.370 | 0.3 |
| Yb–Laa | 2.60 | 7.75 | 0 |
| +10 eq. Hg(II) | 3.80 | 10.98 | 0 |
| Yb–Lbb | 1.76 | 6.31 | 0.2 |
| +10 eq. Hg(II) | 3.85 | 10.60 | 0 |
The effects of selected d-block metal di-cations (CuII, ZnII, CdII and HgII) upon the emission properties of the complexes were probed in buffered aqueous solution (HEPES, pH 7.4). Firstly, the dual emissive nature of the complexes is nicely exemplified through the responses of Eu–La–b. Excitation at 280 nm is not selective for the pyridyl units and therefore results in ligand-centred fluorescence as well as sensitized EuIII emission (5D0 → 7FJ; J = 0, 1, 2, 3 and 4). The emission spectra of Eu–Lb (Fig. 1) show signals originating from pyridyl (ca. 400 nm), anthraquinone (ca. 520 nm) and EuIII (570–720 nm). Addition of CuII partially quenched all of these signals, with EuIII particularly affected. HgII quenched the pyridine fluorescence, enhanced anthraquinone emission and modulated the overall intensity and relative ratios of the EuIII5D0 → 7FJ transitions (including J = 1∶J = 2), suggesting a change in the EuIII coordination sphere. In contrast, addition of either ZnII or CdII selectively enhanced the pyridine-based fluorescence only, suggesting that binding of these ions can occur without inducing a change of coordination environment at EuIII.
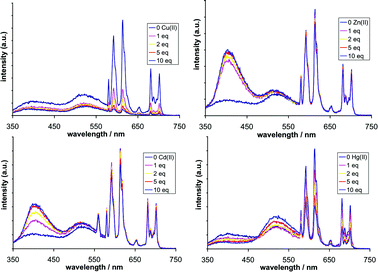 | ||
| Fig. 1 Steady600 dpi in TIF format)??> state emission spectra for Eu–Lb (10−5 M) in the presence (clockwise from top left) of CuII, ZnII, HgII and CdII. Recorded in buffered water (HEPES, pH 7.4) using λex = 280 nm. | ||
The responses of Eu–Laa to MII are shown in the ESI† (Fig. S2). CuII has been shown to quench EuIII emission very effectively, and often by a variety of pathways:15 for Eu–Lb we observe almost complete quenching of the EuIII emission (10 eq. CuII) and a subtle perturbing of ligand-centred fluorescence. In comparison, the addition of HgII resulted in partial quenching of both ligand- and EuIII-centred emission, the latter associated with subtle ratiometric changes, particularly within the J = 4 peaks.
Having established the viability of these luminescent complexes for differentiated metal-ion response, additional studies were undertaken to probe the applicability of these systems to modulated near-IR luminescence output following visible light sensitization. Yb–Laa and Yb–Lb were selected as they demonstrated emission following excitation at 400 nm (i.e. sensitized selectively via the 8-amido-quinoline or 1-amido-9,10-anthraquinone chromophores, respectively). Fig. 2 compares the steady state near-IR emission profiles (2F5/2 → 2F7/2) before and after the addition of HgII (10 eq.). In both cases the intensity of the YbIII-centred emission was dramatically enhanced (up to four-fold in the case of Yb–Laa). UV-vis studies (ESI†, Fig. S3) showed that upon addition of HgII the absorption coefficient at 400 nm increased by only 10%, which does not, therefore, fully account for the enhanced emission.16 Although an improved efficiency of energy transfer (the anthraquinone–YbIII distance may shorten upon HgII binding) could be speculated upon to explain such observations, the increase in emission intensity from YbIII may be attributed to less efficient non-radiative pathways from the 2F5/2 excited state: luminescence lifetimes were obtained in the presence of aqueous HgII and in both complexes showed dramatic extensions beyond >3.5 µs. Companion measurements obtained in D2O showed that, in the presence of HgII, Yb–Laa and Yb–Lb do not possess any inner sphere water (q ≈ 0). These results suggest that although binding of HgII does occur, it may not coordinate to the bridging pyridine unit of Yb–La/ba/b. In fact, the enhancement of YbIII emission and lifetimes suggests that non-radiative deactivation pathways are perturbed upon HgII binding (the integrated emission increases), which may be a consequence of enhanced rigidity within the ligand framework and/or an improved shielding of the YbIII ion from outer sphere solvent effects. In contrast, related measurements on Nd–Lb showed that HgII addition reduced the overall near-IR emission intensity (ESI†, Fig. S4), with a shorter component contributing to the lifetime in water (97 ns), suggesting an increase in inner sphere hydration consistent with the observations on Eu–La–b.8 In all cases these observable changes were found to be reversible, whereby addition of EDTA (ethylenediaminetetraacetic acid) restored the original emission character ([HgEDTA]2−K ≈ 1022).17
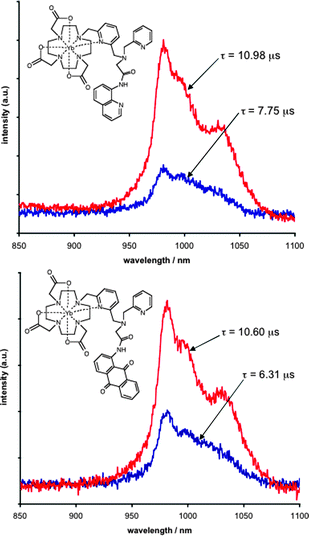 | ||
| Fig. 2 Near-IR steady state emission spectra for 2.5 × 10−4 M Yb–Laa (top) and Yb–Lb (bottom) in the absence (blue) and presence (red) of 10 eq. Hg(ClO4)2. (D2O, HEPES, pH 7.4) using λex = 400 nm. The spectra are annotated with the corresponding luminescence lifetimes. | ||
In summary, this communication describes responsive luminescent probes that demonstrate dual-emission output, visible-light sensitization and modulated near-IR emission output. The unique behaviour of YbIII reveals advantageous sensitivity to di-cation binding within the ligand periphery, rendering it an attractive component for emission lifetime-based near-IR probes. Work is currently underway to elucidate the binding constants and detection limits associated with these near-IR probes.
Cardiff University and EPSRC are thanked for financial support together with the University of Huddersfield. The University of Manchester (Dr Louise Natrajan) is also thanked for provision of elemental analyses and the EPSRC Mass Spectrometry National Service is gratefully acknowledged.
Notes and references
- (a) D. Parker, Coord. Chem. Rev., 2000, 205, 109 CrossRef CAS; (b) C. M. G. dos Santos, A. J. Harte, S. J. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512 CrossRef CAS; (c) J. L. Major and T. J. Meade, Acc. Chem. Res., 2009, 42, 893 CrossRef CAS; (d) G. L. Law, R. Pal, L. O. Palsson, D. Parker and K. L. Wong, Chem. Commun., 2009, 7321 RSC; (e) S. V. Eliseeva and J. C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- (a) V. Fernandez-Moreira, B. Song, V. Sivagnanam, A. S. Chauvin, C. D. B. Vandevyver, M. Gijs, I. Hemmila, H. A. Lehr and J. C. G. Bünzli, Analyst, 2010, 135, 42 RSC; (b) C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS; (c) B. McMahon, P. Mauer, C. P. McCoy, T. C. Lee and T. Gunnlaugsson, J. Am. Chem. Soc., 2009, 131, 17542 CrossRef CAS.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512 RSC.
- (a) J. L. Major, G. Parigi, C. Luchinat and T. J. Meade, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13881 CrossRef CAS; (b) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517 CrossRef CAS.
- S. Faulkner, S. J. A. Pope and B. P. Burton-Pye, Appl. Spectrosc. Rev., 2005, 40, 1 CrossRef CAS.
- M. Andrews, A. J. Amoroso, L. P. Harding and S. J. A. Pope, Dalton Trans., 2010, 39, 3407 RSC.
- A. Dadbhoy, S. Faulkner and P. G. Sammes, J. Chem. Soc., Perkin Trans. 2, 2002, 348 RSC.
- (a) S. J. A. Pope and R. H. Laye, Dalton Trans., 2006, 3108 RSC; (b) S. J. A. Pope, V. Boote, A. M. Kenwright and S. Faulkner, J. Chem. Soc., Dalton Trans., 2003, 3780 RSC.
- (a) J. E. Jones and S. J. A. Pope, Dalton Trans., 2009, 8421 RSC; (b) P. Dahiya, M. Kumbhakar, D. K. Maity, T. Mukherjee, J. P. Mittal, A. B. R. Tripathi, N. Chattopadhyay and H. Pal, Photochem. Photobiol. Sci., 2005, 4, 100 RSC.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- E. L. Que, E. Gianolio, S. L. Baker, S. Aime and C. J. Chang, Dalton Trans., 2010, 39, 469 RSC.
- N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. Bünzli, Inorg. Chem., 2009, 48, 7937 CrossRef CAS; N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. Bünzli, Inorg. Chem., 2009, 48, 2908 CrossRef CAS.
- M. H. V. Werts, R. T. F. Jukes and J. W. Verhoeven, Phys. Chem. Chem. Phys., 2002, 4, 1542 RSC.
- S. Faulkner, M.-C. Carrie, S. J. A. Pope, J. Squire, A. Beeby and P. G. Sammes, Dalton Trans., 2004, 1405 RSC.
- (a) M. A. Kessler, Anal. Chim. Acta, 1998, 364, 125 CrossRef CAS; (b) T. Gunnlaugsson, J. P. Leonard, K. Senechal and A. J. Harte, Chem. Commun., 2004, 782 RSC.
- J. B. Coldwell, C. E. Felton, L. P. Harding, R. Moon, S. J. A. Pope and C. R. Rice, Chem. Commun., 2006, 5048 RSC.
- S. M. Miller, D. P. Ballou, V. Massey, C. H. Williams, Jr. and C. T. Walsh, J. Biol. Chem., 1986, 261, 8081 CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic characterisation and additional photophysical data. See DOI: 10.1039/c0cc00210k |
| This journal is © The Royal Society of Chemistry 2011 |