An efficient oxidative dearomatization–radical cyclization approach to symmetrically substituted bicyclic guttiferone natural products†‡
Nicholas A.
McGrath
,
Joshua R.
Binner
,
Georgios
Markopoulos
,
Matthew
Brichacek
and
Jon T.
Njardarson
*
Department of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, NY, USA. E-mail: jn96@cornell.edu; Fax: +1 607 255 4137; Tel: +1 607 254 5035
First published on 7th July 2010
Abstract
Detailed in this communication is an efficient synthetic approach towards the guttiferone family of natural products. Oxidatively unraveling a para-quinone monoketal followed by consecutive 5-exo radical cyclizations provides the bicyclic core. An additional strength of this approach is a late stage asymmetric desymmetrization of an advanced symmetric intermediate.
In recent years a vast number of bridged bicyclic polyprenylated acylphloroglucinol natural products have been reported.1 Perhaps the most famous member of this family of natural products is hyperforin, which is one of the main chemical constituents of the commonly used natural remedy St. John's wort and has in recent years shown promise as a potential anticancer agent.2 We have in the last few years established a research program focused on synthesizing and evaluating unique bridged bicyclic natural product anti cancer agents. The bridged phloroglucinol family drew our attention early on, but the catalyst for launching a synthetic program towards their synthesis was a report detailing the sirtuin inhibitory activity of hyperforin and guttiferone G (Fig. 1).3 The sirtuins are considered high value targets for developing new anticancer agents and gaining more insight into improving longevity.4 Not surprisingly, there is great interest in finding small molecule inhibitors, which selectively block the function of any of the seven known enzymes of the sirtuin family (SIRT1-7).5
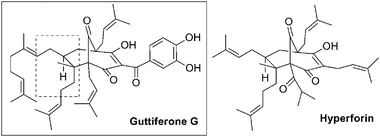 | ||
| Fig. 1 Guttiferone G and hyperforin. | ||
Hyperforin and guttiferone G share many structural similarities in addition to the common bridged bicyclic trione core, neither natural product has been synthesized to date.6 In our minds the most attractive difference is the bis-prenyl bridgehead substitution of guttiferone G, which means that the fully substituted trione part of the molecule is symmetrical.
This local symmetry opens the door for exciting synthetic designs and more importantly for late introduction of chirality. By inspecting the literature for other such structures we chose to limit our search only to compounds containing stereocenters at both C5 and C6, similar to that found in both guttiferone G and hyperforin. In doing so we excluded a fair number of symmetrically substituted compounds containing the more common C5 gem-dimethyl substitution pattern. Our search revealed that eight guttiferones sharing a symmetrical [3.3.1] bridged bicyclic core had been reported in the last twenty years since the first such member was reported (guttiferone A).7 The only structural differences between members of this remarkable family are the substitution of two adjacent stereocenters (C5 and C6), minor variations with respect to the degree of oxidation of the benzoyl group and the absolute configuration of the desymmetrized core (Fig. 2).
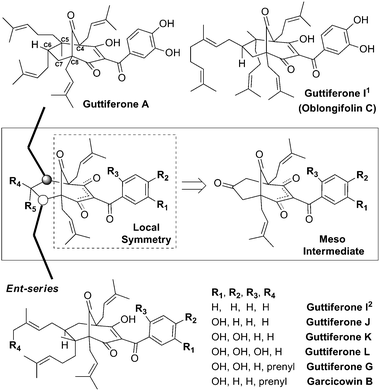 | ||
| Fig. 2 Guttiferones containing locally symmetrical bicyclic cores. | ||
A closer look at the published data for these eight compounds reveals that although there are four possible arrangements for each C5/C6 substitution pattern for each enantiomeric series, it seems nature always prefers to place the two large groups (geranyl and prenyl) trans to each other. Guttiferone A is the only member of this natural product stereochemical library whose absolute configuration has been unambiguously established.8 The data suggest that guttiferone A and I1 belong to the same enantiomeric series9 and that the other six (guttiferones I2, J, K, L, G and garcicowin B) natural products belong to the opposite one.10 The latter six structures are remarkably similar, differing only in oxygenation of the benzoyl group and whether there is a prenyl or geranyl group at the C6-position.
Although this unique natural product collection has never been tested as one, each member has been shown to exhibit promising anti-cancer activity ranging from general cytotoxicity10,11 to potential as protease inhibitors,12 antiapoptotic13 or antiproliferative agents.10 In addition, several studies have been reported on their various specific biological functions beyond cancer.9,14 As stated above, we are most excited about the sirtuin inhibitory activity of guttiferone G3 and to learn how the other seven members of this family compare. Such a study would provide important SAR clues on the relative importance of the C5, C6 or benzoyl substitutions on sirtuin inhibition.
Our retrosynthetic analysis is presented in Scheme 1. It relies on the late stage desymmetrization of 2, which we envision could be converted to all eight targeted natural products by proper choice of reagents. This intermediate is rapidly accessed via tandem 5-exo radical cyclizations (3) enabled by oxidative dearomatization of a strategically functionalized para-hydroquinone, which in turn is assembled from phenol 415 and malonate derivative 5.
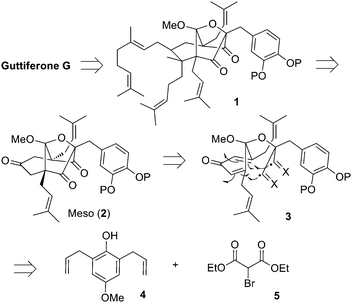 | ||
| Scheme 1 Guttiferone G retrosynthesis. | ||
Our synthetic efforts commenced with known diallyl ether 4, which is readily accessible from 4-methoxy phenol (Scheme 2). The free phenol was alkylated with diethyl 2-bromomalonate (5) to afford 6. The esters were converted into bromomethyl groups (7) following standard procedures. Selective deprotection of the methyl capped phenol was accomplished using BCl3 and hypervalent iodine mediated oxidative dearomatization yielded the desired dienone acetal radical cyclization precursor 8. Despite discouraging literature precedents,16 which suggested preferential formation of 9 over 10 we decided to test the tandem 5-exo/5-exo radical cyclization thesis. Cyclization proceeded smoothly and selectively, affording only ketal 9 and not even trace amounts of bridged bicyclic ketal 10. We argued that placing a large group in between the two radical sites would force the two radicals to be on the same face, which is critical for accessing the bicyclic motif. This logic flows well with our synthetic design because an oxygenated phenacyl group happens to reside in this exact position on the guttiferones. Towards that end, diester6 was alkylated (11) and reduced to diol12. Bromination of the bis-neopentyl alcohols was accomplished in two steps to give 13. Selective deprotection of the methyl ether was eventually accomplished using B(C6F5)3 in the presence of triethylsilane.17
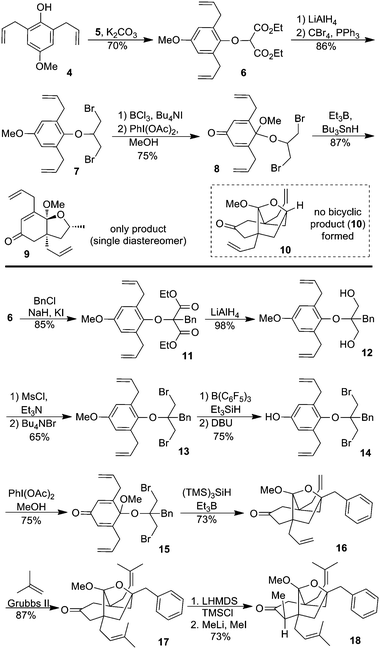 | ||
| Scheme 2 Synthesis of the bridged bicyclic core of the guttiferones. | ||
Dearomatization proceeded uneventfully to produce ketal 15, which gratifyingly cyclized to form the desired symmetrical bridged bicyclic product 16 as the only product. Cross metathesis of 16 with 2-methyl propene gave bis-prenylated 17.18 To test the facial selectivity in the alkylation of the bicyclic ketone17, the enolate generated with LHMDS was trapped as the silyl ether. Treatment with MeLi and MeI resulted in exclusive methylation from the exo-face (18). This stereoselective alkylation lends promise to our stated goal of being able to access any member of this natural product class by changing the order of the alkylation sequence.
In order to explain the profound reversal of selectivity in the cyclization of 8 and 15, we performed density functional theory calculations (UB3LYP 6-31G(d)). The two possible transition states for the first radical cyclization were found and the more stable in each case (21 and 23) is depicted in Scheme 3. We see that in the case of R = H, the transition state having the methylene bromide on the exo face is preferred by 2.8 kcal mol−1. Alternatively with R = Bn, steric interactions with the benzyl group force it to the less hindered exo face and the methylene bromide to the endo which is competent for further cyclization.
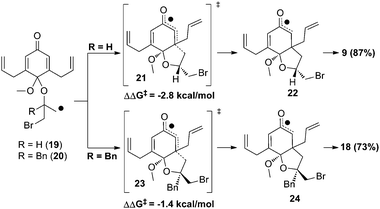 | ||
| Scheme 3 Rationalization of stereochemical outcome. | ||
In order to assess our ability to carry out a late-stage desymmetrization to access either enantiomeric series of the guttiferone family, we employed chiral amide bases (Table 1). The asymmetry in the deprotonation was determined by trapping the enolate as a Mosher ester. Both enantiomers of the enolate were trapped to give the corresponding diastereomeric Mosher esters. An increased selectivity was observed with LiCl being added prior to deprotonation.19
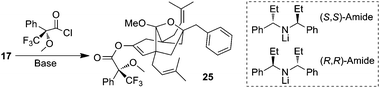
| Base | Additive | Temp (°C) | Yield (%) | dr |
|---|---|---|---|---|
| LHMDS | — | −78 | 85 | 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 ∶1 |
| (R,R)-Amide | — | −78 | 87 | 3∶1 |
| (R,R)-Amide | LiCl | −78 | 83 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) ∶1 ∶1
|
| (S,S)-Amide | LiCl | −78 | 75 | 1∶6 |
| (R,R)-Amide | LiCl | −100 | 72 | 3∶1 |
In conclusion, we have developed an efficient approach to this exciting class of natural products that takes advantage of the inherent local symmetry present in the bicyclic structure. The complex core was accessed by employing a unique double radical cyclization of a p-quinone ketal derived from a simple aromatic precursor. The shape of the molecule controls the facial selectivity during the ketone alkylation and the use of a chiral amide base provides access to either enantiomeric series.
We gratefully acknowledge the NIH for a CBI training grant (T32GM008500 to NAM and MB), NIH-NIGMS (RO1 GM086584) and most recently an ARRA administrative supplement to RO1 GM086584.
Notes and references
- R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963 CrossRef CAS; I. P. Singh and S. B. Bharate, Nat. Prod. Rep., 2006, 23, 558 RSC; U. M. Acuna, N. Jancovski and E. J. Kennelly, Curr. Top. Med. Chem., 2009, 9, 1560 CrossRef CAS.
- M. Dona, I. Dell'Aica, E. Pezzato, L. Sartor, F. Calabrese, M. D. Barbera, A. Donella-Deana, G. Appendino, A. Borsarini, R. Caniato and S. Garbisa, Cancer Res., 2004, 64, 6225 CrossRef CAS; B. Martinez-Poveda, A. R. Quesada and M. A. Medina, Int. J. Cancer, 2005, 117, 775 CrossRef CAS; C. Quiney, C. Billard, C. Salanoubat, J. D. Fourneron and J. P. Kolb, Leukemia, 2006, 20, 1519 CrossRef CAS; M. Rothley, A. Schmid, W. Thiele, V. Schacht, D. Plaumann, M. Gartner, A. Yektaoglu, F. Bruyere, A. Noel, A. Giannis and J. P. Sleeman, Int. J. Cancer, 2009, 125, 34 CrossRef CAS.
- C. Gey, L. H. Kyrylenko, L.-H. D. Nguyen, A. Buttner, H. D. Pham and A. Giannis, Angew. Chem., Int. Ed., 2007, 46, 5219 CrossRef CAS.
- L. R. Saunders and E. Verdin, Oncogene, 2007, 26, 5489 CrossRef CAS; C. L. Brooks and W. Gu, Nat. Rev. Cancer, 2009, 9, 123 CrossRef CAS.
- J. C. Milne and J. M. Denu, Curr. Opin. Chem. Biol., 2008, 12, 11 CrossRef CAS; A. Mai, D. Cheng, M. T. Bedford, S. Valente, A. Nebbioso, A. Perrone, G. Brosch, G. Sbardella, F. D. Bellis, M. Miceli and L. Altucci, J. Med. Chem., 2008, 51, 2279 CrossRef CAS; F. J. Alcain and J. M. Villalba, Expert Opin. Ther. Pat., 2009, 19, 283 Search PubMed.
- Ent-hyperforin: Y. Shimizu, S.-L. Shi, H. Usuda, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2010, 49, 1103 Search PubMed.
- K. R. Gustafson, J. W. Blunt, M. H. G. Munroe, R. W. Fuller, T. C. McKee, J. H. Cardellina, J. B. McMahon, G. M. Gragg and M. R. Boyd, Tetrahedron, 1992, 48, 10093 CrossRef CAS.
- B. N. Lenta, D. T. Nougoue, K. P. Devkota, P. A. Fokou, S. Ngouela, E. Tsamo and N. Sewald, Acta Crystallogr., Sect. E, 2007, 63, 01282; F. T. Martins, J. W. Cruz, P. B. M. C. Derogis, M. H. Dos Santos, M. P. Veloso, J. Ellen and A. Doriguetto, J. Braz. Chem. Soc., 2007, 18, 1515 CAS.
- K. Herath, H. Jayasuriya, J. G. Ondeyka, Z. Guan, R. P. Borris, E. Stijfhoorn, D. Stevenson, J. Wang, N. Sharma, K. MacNaul, J. G. Menke, A. Ali, M. J. Schulman and S. B. Singh, J. Nat. Prod., 2005, 68, 617 CrossRef CAS; W. Hamed, S. Brajeul, F. Mahuteau-Betzer, O. Thoison, S. Mons, B. Delpech, N. V. Hung, T. Sevenet and C. Marazano, J. Nat. Prod., 2006, 69, 774 CrossRef CAS.
- R. B. Williams, J. Hoch, T. E. Glass, R. Evans, J. S. Miller, J. H. Wisse and D. G. I. Kingston, Planta Med., 2003, 69, 864 CrossRef CAS; J. Merza, S. Mallet, M. Litaudon, V. Dumontet, D. Seraphin and P. Richomme, Planta Med., 2006, 72, 87 CAS; S. Cao, P. J. Brodie, J. S. Miller, F. Ratovoson, C. Birkinshaw, S. Randrianasolo, E. Rakotobe, V. E. Rasamison and D. G. I. Kingston, J. Nat. Prod., 2007, 70, 686 CrossRef CAS; G. Xu, W. L. T. Kan, Y. Zhou, J.-Z. Song, Q.-B. Han, C.-F. Qiao, C.-H. Cho, J. A. Rudd, G. Lin and H.-X. Xu, J. Nat. Prod., 2010, 73, 104 CrossRef CAS.
- P. Protiva, M. E. Hopkins, S. Baggett, H. Yang, M. Lipkin, P. R. Holt, E. J. Kennelly and W. I. Bernard, Int. J. Cancer, 2008, 123, 687 CrossRef CAS.
- F. T. Martins, D. M. Assis, M. H. Santos, I. Camps, M. P. Veloso, M. A. Juliano, L. C. Alves and A. C. Doriguetto, Eur. J. Med. Chem., 2009, 44, 1230 CrossRef CAS.
- G. Xu, C. Feng, Y. Zhou, Q.-B. Han, C.-F. Qiao, S.-X. Huang, D. C. Chang, Q.-S. Zhao, K. Q. Luo and H.-X. Xu, J. Agric. Food Chem., 2008, 56, 11144 CrossRef CAS; S.-X. Huang, C. Feng, Y. Zhou, G. Xu, Q.-B. Han, C.-F. Qiao, D. C. Chang, K. Q. Luo and H.-X. Xu, J. Nat. Prod., 2009, 72, 130 CrossRef CAS.
- F. J. Naldoni, A. L. R. Claudino, J. W. Cruz, J. K. Chavasco, P. M. F. e Silva, M. P. Veloso and M. H. dos Santos, J. Med. Food, 2009, 12, 403 CrossRef CAS; J. Kolodziejczyk, M. Masullo, B. Olas, S. Piacente and B. Wachowicz, Platelets, 2009, 20, 487 CrossRef CAS; I. O. Pereira, M. J. Margues, A. L. R. Pavan, B. S. Codonho, C. L. Barbieri, L. A. Beijo, A. C. Doriguetto, E. C. D'Martin and M. H. dos Santos, Phytomedicine, 2010, 17, 339 CrossRef CAS.
- F.-T. Hong, K.-S. Lee, Y.-F. Tsai and C.-C. Liao, J. Chin. Chem. Soc., 1998, 45, 1 CAS.
- F. Villar, O. Eguey and P. Renaud, Org. Lett., 2000, 2, 1061 CrossRef CAS.
- V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179 CrossRef CAS.
- A. K. Chatterjee, D. P. Sanders and R. H. Grubbs, Org. Lett., 2002, 4, 1939 CrossRef CAS.
- P. O'Brien, J. Chem. Soc., Perkin Trans. 1, 1998,(8), 1439 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and spectroscopic data. See DOI: 10.1039/c0cc01419b |
| ‡ This article is part of the ‘Emerging Investigators’ themed issue for ChemComm |
| This journal is © The Royal Society of Chemistry 2011 |