Preconcentration of selenium by living bacteria immobilized on silica for microwave induced plasma optical emission spectrometry with continuous powder introduction
Anna
Tyburska
and
Krzysztof
Jankowski
*
Warsaw University of Technology, Faculty of Chemistry, Department of Analytical Chemistry, 00-664, Warszawa, ul. Noakowskiego 3, Poland. E-mail: kj@ch.pw.edu.pl
First published on 28th January 2011
Abstract
The analytical potential of living bacteria Lactobacillus plantarum for the preconcentration of selenium from mineral water and beer samples followed by the selenium determination by continuous powder introduction microwave induced plasma-optical emission spectrometry (CPI-MIP-OES) has been investigated. The use of the bacteria immobilized on silica permits on-column preconcentration of selenium with minimum sample pretreatment by the pH adjustment. Experimental conditions for the preconcentration were optimized and the mechanism of the biosorption was investigated. Results showed that the use of the preconcentration stage leads to the substantial lowering of the detection limit of selenium by OES. The detection limit of 52 ng g−1 by the CPI-MIP-OES was achieved which corresponds to 0.06 ng mL−1 in the sample solution regarding the preconcentration factor of 1000. Precision of 5 replicate measurements expressed as relative standard deviation was about 3%. The utility of the method was demonstrated in the determination of selenium in hard drinking water ERM-CA011a as well as in mineral water and beer samples.
Introduction
The determination and speciation of selenium in environmental and food samples are of great importance, due to the biological effects, including deficiency and intoxication effects, which depend highly on its concentration and chemical form. Spectrometric techniques are widely used for the determination of selenium in a variety of environmental samples. However, for a number of samples including waters and beverages the selenium content is too low to permit its accurate determination without preconcentration stage. In recent years solid phase extraction (SPE) has gained a widespread acceptance as a preconcentration technique. Usually, the analyte retention on the sorbent is followed by its elution with a portion of liquid or by thermal desorption and transport of a vapour to the atomization/radiation source. The alternative procedure is the direct introduction of preconcentrated analyte as analyte-on-sorbent particles into the low gas flow microwave induced plasma. The analytical performance of SPE combined with continuous powder introduction microwave induced plasma optical emission spectrometry (CPI-MIP-OES) has been discussed elsewhere.1–3Both inorganic and organic sorbents were used for the preconcentration of selenium.4,5 In the last decade living organisms such as algae, yeast, fungi and bacteria were examined for the purpose of trace metal preconcentration. A number of microorganisms have been identified showing very strong adsorption affinities toward Hg, Pd, Cd, Cu, and Se.6–15 The possible advantage of using microorganisms is that the cell wall has many constituents that can be active in metal binding. The metal uptake by biomass can take place by an active mode called bioaccumulation (transport into the cell with subsequent transformation) or by passive mode termed as biosorption.7 The mechanism responsible for metal biosorption may include ionic interaction and complex formation between metal ions and biomass. It was observed that dead cells accumulate heavy metals to an equal or greater extent than living cells.
Both free and immobilized microorganisms could be used as biosorbents. The immobilization of microorganisms offers many advantages including better process stability and the possibility of use in the on-column procedure. A few immobilization techniques are known including the immobilization on the support surface or the encapsulation inside the organic matrix. Several supports have been used for immobilization purposes, while silica gel,14,15 controlled pore glass16 and alginate7 are the most widely used in analytical procedures. Excellent biosorbents for the preconcentration of selenium are some bacteria,11–13 yeast, particularly baker's yeast Saccharomyces cerevisiae,9 and algae.10 Although the bacteria Lactobacillus plantarum proved to be a very good biosorbent for selenium,17,18 their usefulness for the analytical preconcentration of selenium has not been described yet. In this work, the bacteria immobilized on silica were employed for selenium preconcentration. The analytical potential of the CPI-MIP-OES coupled with selenium preconcentration is demonstrated in the analysis of mineral water and beer samples and hard drinking water ERM-CA011a.
Experimental
Reagents and materials
High grade analytical reagent chemicals were employed in the preparation of all solutions. Standard solution of selenium(IV) (100 mg L−1) was prepared by dissolution of suitable amount of sodium hydrogen selenite (POCH, Poland) in a small volume of nitric acid, followed by dilution with MILLI-Q purified water. All experiments described in this work were carried out with the use of Se(IV) solution unless otherwise stated.Standard solution of selenium(VI) (100 mg L−1) was prepared by dissolution of suitable amount of ammonium selenate (POCH, Poland) in a small volume of nitric acid, followed by dilution with MILLI-Q purified water.
Standard solution of organic selenium (86 mg L−1) was prepared by dissolution of suitable amount of selenomethionine (Sigma-Aldrich) in water.
Stock solutions of concomitant elements (100 mg L−1) were prepared from pure nitrate or chloride salts.
Silica gel of chromatographic grade, particle size 32–63 µm, was used.
The bacteria L. plantarum were purchased in a local pharmacy. One capsule (540 mg) of the pharmaceutical preparation contains 1 billion cells of three strains of lyophilized bacteria L. plantarum and 132 mg of inulin.
Apparatus
Selenium present in aqueous solutions was determined by ICP-OES with pneumatic nebulization of solutions. The ICP-OES spectrometer (Integra XL, GBC Australia) with radial viewing was equipped with the TR50C1 (Meinhard, USA) concentric nebulizer. The ICP operating conditions are given in Table 1.| He-MIP-OES | Ar-ICP-OES | |
|---|---|---|
| Incident power/W | 350 | 1200 |
| Viewing mode | Axial | Radial |
| Height above coil/mm | — | 4 |
| Intermediate gas flow rate/L min−1 | — | 0.3–0.5 |
| Plasma/outer gas flow rate/L min−1 | 0.2 | 12.0 |
| Sampling/nebulising gas flow rate/L min−1 | 0.4 | 0.5 |
| Wavelength/nm | Se 196.03 | |
| Se 203.99 | ||
The measurements of solid samples were made with the use of the MIP spectrometer (MIP 750MV, Analab, Poland) equipped with TEM microwave cavity. Continuous introduction of the powdered samples was achieved by passing a low flow of helium (up to 200 mL min−1) through the sample chamber. A suitable powder loading was about 0.5 g with particle sizes between 32 and 63 µm. A schematic diagram of the CPI system is shown in Fig. 1. The MIP operating conditions are given in Table 1.
 | ||
| Fig. 1 Schematic diagram of the continuous powder introduction system. | ||
Immobilization procedure
Silica is a good substrate for immobilization because of its chemical stability, noncompressibility, and availability in a variety of sizes and porosities. The bacteria were immobilized using the method developed by Mahan and Holcombe for algae cells.15 Approximately 500 mg of the lyophilized bacteria L. plantarum was mixed with 2 g of silica (32–63 µm). The blend was wetted with a minimal amount of water and thoroughly mixed. The resulting paste was dried. The last two steps were repeated twice to improve the immobilization efficiency. The silica–bacteria briquette was sieved to remove particles smaller than 32 µm. This immobilization procedure, based on the adsorption process, increases the resistance of the substrate preventing microbial leaching from the column. Electron micrograph of bacteria L. plantarum immobilized on silica is presented in Fig.2.
Preconcentration procedure
A polypropylene column of 6 mm i.d. was loaded with 0.5 g of dried L. plantarum immobilized on silica. Then the sorbent material was wetted with water. The pH of the sample was adjusted to about 5 using diluted HCl and the sample passed through the column at a flow rate of several mL min−1. The column was washed with 5 mL of water. Then the column was unloaded, the biosorbent was dried, and the selenium was subsequently determined by CPI-MIP-OES. The whole analytical procedure takes 5–12 hours depending on the sample volume ranging from 100 mL to 1.5 L.Measurement procedure
A dried sample obtained during the preconcentration procedure was placed in the sample chamber of the CPI system. The helium plasma was ignited and the microwave power was adjusted to 350 W. Then, both the sampling gas flow and plasma gas flow were adjusted to 400 and 200 mL min−1, respectively. After stabilization of the signal the selenium emission was measured at 1960.26 and 2039.85 A (Fig. 3). The measurements were made in the sequential mode with off peak background correction. | ||
| Fig. 3 Spectrum of helium MIP with the analyte-on-biosorbent powder introduction. | ||
Results and discussion
Optimization of the Se biosorption conditions
Experimental conditions were optimized to attain maximum% sorption of selenium. A portion of 0.5 g of the sorbent was introduced into the 25 mL of solution containing 1 µg mL−1 of selenium. The sorption efficiency was calculated on the basis of selenium content in the sorbent which was determined by CPI-MIP-OES. The operating parameters studied were as follows: pH ranged from 3 to 9, temperature ranged from 20 °C to 50 °C, and sorption time ranged from 1 to 48 hours. As shown in Fig. 4, the most effective sorption of selenium was in the range of 4–6 pH. The higher pH value is not recommended, because of the decrease of the biological activity of bacteria in neutral or alkaline medium. In the range examined the biosorption of selenium is not affected by temperature. Again, operating at temperature higher than 50 °C is inappropriate, because the bacteria L. plantarum lose their biological activity. Thus, the biosorption was carried out at room temperature. Under such conditions the influence of sorption time was examined. As shown in Fig. 5 satisfactory% sorption is observed after 1.5 h. In order to support the above mentioned experimental results the mass balance of biosorption process for 15 µg of selenium was provided. The selenium content in the biosorbent was found to be 14.2 µg by CPI-MIP-OES while 0.6 µg of Se was determined in the supernatant using ICP-OES with solution nebulization. Thus, above 98% of the selenium introduced was recovered in both phases after biosorption.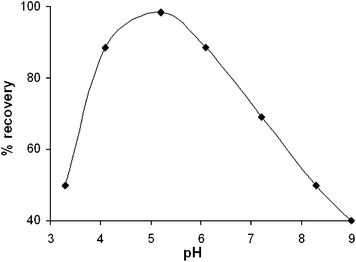 | ||
| Fig. 4 Effect of pH on selenium uptake by L. plantarum immobilized on silica: biosorbent mass 0.5 g, sorption time 1.5 hour, 25 °C, selenium concentration 1 µg mL−1. | ||
 | ||
| Fig. 5 Effect of sorption time on selenium uptake by L. plantarum immobilized on silica: biosorbent mass 0.5 g, pH 5, 25 °C, selenium concentration 1 µg mL−1. | ||
Batch and on-column preconcentration
In order to determine the sorption capacity of bacteria L. plantarum immobilized on silica, 1 g of the biosorbent was added to solutions containing the increasing amount of Se at pH 5 at room temperature and the sorption time was 1.5 h. Under such conditions the sorption capacity was 150 µg g−1. However, it was about 700 µg g−1 with respect to the biomass taking into account that only a small fraction of selenium was retained on silica (see below). This value of the sorption capacity is comparable with those reported by others for various bacteria strains.17In the batch procedure 1 gram of the biosorbent was added to the 0.05–1 L of solution (pH 5) containing 10 µg of Se. Next the mixture was continuously stirred for 1.5 hour. Then, after taking off the Se-biosorbent solid phase by decantation and centrifugation it was washed with water and again centrifuged. Next the biosorbent was dried and sieved to meet CPI technique requirements, and the selenium was subsequently determined by CPI-MIP-OES. The recovery studies have shown quantitative retention of selenium (95–100%) for sample volumes up to 1 L. This allows attaining high preconcentration factor of about 1000.
The biosorbent was applied for on-column preconcentration of selenium. The columns (6 mm i.d.) were packed with a known weight (0.5 g) of the biosorbent. 50 mL of selenium solution (1 µg mL−1) at pH 5 was passed through the column at a flow rate of 1 mL min−1. The selenium content in the effluent was determined by ICP-OES. Then the column was washed with deionized water, the biosorbent was dried and the selenium was determined by CPI-MIP-OES. The recovery of Se was in the range 95–100%. Similar recovery was obtained when 1 L of solution containing 10 µg of Se was passed through the column.
Mechanism of biosorption of selenium
In order to establish a possible mechanism of selenium sorption three batch processes were carried out simultaneously. In each case 1 g of sorbent was put into the solution containing 15 µg of Se(IV). However, in the first process living bacteria immobilized on silica were used, while in the second the biosorbent was previously exposed to UV radiation for 20 min for sterilization. In the third case the sorption of selenium on silica was examined. After 1.5 hour of stirring, each sorbent was centrifuged and dried, and the selenium was subsequently determined by CPI-MIP-OES. The average amount of selenium determined in each case was 14.2, 10.0 and 0.6 µg, respectively. The comparison of the results shows that about 66% of Se is adsorbed on the external surface of the cell wall, about 30% is accumulated by living bacteria inside the cells and approximately 4% is adsorbed on the silica support under experimental conditions. Moreover, it was tested that selenium(VI) and selenomethionine are not accumulated by the bacteria.Analytical performance
The limit of detection (3σ) of selenium at 203.9 nm by CPI-MIP-OES technique was calculated to be 52 ng g−1 while for aqueous solution it was 0.06 µg L−1 assuming the preconcentration factor of 1000. The precision of 3.4% was calculated from the RSD of the mean of five replicate measurements of analyte standard using a mass 100-fold above the LOD. The method of determination of selenium by external calibration at wavelength 203.9 nm using CPI-MIP-OES in combination with the preconcentration of selenium by bacteria L. plantarum immobilized on silica was worked out. A set of calibration standards covering the concentration range of 0.2–150 µg of Se per 1 g of the biosorbent was prepared by biosorption of the analyte under experimental conditions described above. The calibration graph was linear in the range from 0.25 to 100 µg g−1 with the correlation coefficient (R2) of 0.9996.The effect of 6 concomitant ions on the sorption and determination of selenium was examined and results indicated that no interference was observed when 250 µg or 2.5 mg As, Sb, Fe, Cu, Na and Ca was added together with 25 µg of Se in 100 mL of solution (Table 2). The concomitant to Se concentration ratios for Fe, Cu, Na and Ca generally reflect an actual presence of these elements in a hard drinking water and beer samples analyzed. The recovery of selenium varied from 97 to 102%.
| Ion | [cM]/[cSe] | Se recovery (%) |
|---|---|---|
| Fe2+ | 10 | 98 |
| Fe2+ | 100 | 97 |
| Na+ | 10 | 100 |
| Na+ | 100 | 101 |
| Sb3+ | 10 | 99 |
| Sb3+ | 100 | 98 |
| As3+ | 10 | 101 |
| As3+ | 100 | 99 |
| Ca2+ | 10 | 99 |
| Ca2+ | 100 | 99 |
| Cu2+ | 10 | 100 |
| Cu2+ | 100 | 102 |
Determination of selenium in water and beer
The utility of the method was demonstrated by the determination of selenium in mineral water, beer, and wort. The determination of selenium in mineral water and beer by MIP-OES or ICP-OES requires analyte preconcentration because the selenium content is below DL values offered by these techniques. The original pH of the samples varied from 6.5 to 7. After pH adjustment to 5–5.5 the samples of 1.5 L were transferred at the flow rate of about 3 mL min−1 through the column loaded with 0.5 g of the biosorbent that led to about 3000 preconcentration factor. Next, the biosorbent was washed with 5 mL of water and dried, and selenium retained on the biosorbent was subsequently determined by CPI-MIP-OES.Two calibration methodologies were evaluated for determination of selenium by CPI-MIP-OES. The method of standard addition consisted of the addition of the known amount of selenium using a weight of selenium yeast Selm-1 certified reference material (containing 2059 mg kg−1 of selenium) directly to the dried analyte-on-biosorbent sample. The preparation was mixed thoroughly to ensure their homogeneity. The second option was external calibration with powdered standards obtained by biosorption from selenium standard solutions and drying of resulted materials. The results are presented in Table 3. Both calibration methodologies offer similar accuracy and precision.
The selenium concentration determined in the mineral water is close to that declared by the manufacturer and also this determined previously by direct hydride generation ICP-OES.19 The selenium concentration in beer “Lech” was similar to that obtained by hydride generation ETAAS.20 The analytical viability of the analytical procedure was examined by the determination of selenium in hard drinking water ERM-CA011a. The determination of selenium in this water sample as well as in wort required much lower preconcentration rate due to the relatively high content of selenium. The concentration found is in good agreement with the respective certified value.
Conclusion
Trace selenium can be successfully determined by CPI-MIP-OES at low ppb levels. The use of the on-column biosorption by bacteria L. plantarum immobilized on silica for the preconcentration of selenium results in improved detection power. Both external calibration and standard addition methodologies are suitable for the quantitative determination of selenium in real samples. Alkali and transition metals present in mineral and drinking water do not interfere with the determination of selenium as well as the organic matrix present in beer and wort does not affect the preconcentration and the determination of selenium. Both the high preconcentration factor available for sorption technique and good sensitivity of optical emission spectrometry technique with helium plasma enable the determination of selenium in real samples at concentrations down to 1 ng mL−1.Acknowledgements
This work was financially supported by Warsaw University of Technology.References
- K. Jankowski and A. Jackowska, Trends Appl. Spectrosc., 2007, 6, 17–25 Search PubMed.
- K. Jankowski and E. Reszke, Microwave Induced Plasma Analytical Spectrometry, RSC Publishing, 2010 Search PubMed.
- K. Jankowski, J. Yao, K. Kasiura, A. Jackowska and A. Sieradzka, Spectrochim. Acta, Part B, 2005, 60, 369–375 CrossRef.
- K. Pyrzyńska, P. Drzewicz and M. Trojanowicz, Anal. Chim. Acta, 1998, 363, 141–146 CrossRef CAS.
- V. Camel, Spectrochim. Acta, Part B, 2003, 58, 1177–1233 CrossRef.
- A. Nakajima and T. Sakaguchi, Appl. Microbiol. Biotechnol., 1986, 24, 59–64 CAS.
- B. Godlewska-Żyłkiewicz, CRC Crit. Rev. Anal. Chem., 2001, 31, 175–189 CrossRef CAS.
- A. Vecchio, C. Finoli, D. Di Simine and V. Andreoni, Fresenius' J. Anal. Chem., 1998, 361, 338–342 CrossRef CAS.
- T. Perez-Corona, Y. Madrid and C. Camara, Anal. Chim. Acta, 1997, 345, 249–255 CrossRef CAS.
- L. Shunxin, Q. Shahua, H. Ganquan and H. Fei, Fresenius' J. Anal. Chem., 1999, 365, 469–471 CrossRef CAS.
- J. Rivera-Utrilla, J. Bautista-Toledo, M. A. Ferro-Garcia and C. Moreno-Castilla, Carbon, 2003, 41, 323–330 CrossRef CAS.
- L. C. Robles, B. de Celis, J. M. Lumbereras and A. J. Aller, Anal. Commun., 1997, 34, 409–411 RSC.
- A. J. Aller and L. C. Robles, J. Anal. At. Spectrom., 1998, 13, 469–476 RSC.
- T. Perez-Corona, Y. Madrid-Albarran, C. Camara and E. Beceiro, Spectrochim. Acta, Part B, 1998, 53, 321–329 CrossRef.
- C. A. Mahan and J. A. Holcombe, Anal. Chem., 1992, 64, 1933–1939 CrossRef CAS.
- A. Maquieira, H. Elmahadi and R. Puchades, J. Anal. At. Spectrom., 1996, 11, 99–106 RSC.
- M. Calomme, J. Hu, K. Van Den Branden and D. A. Vanden Berghe, Biol. Trace Elem. Res., 1995, 47, 379–383 CrossRef CAS.
- S. K. Xia, L. Chen and J. Q. Liang, J. Agric. Food Chem., 2007, 55, 2413–2417 CrossRef CAS.
- A. Tyburska, K. Jankowski, A. Ramsza, E. Reszke, M. Strzelec and A. Andrzejczuk, J. Anal. At. Spectrom., 2010, 25, 210–214 RSC.
- H. Matusiewicz and M. Mikołajczak, J. Anal. At. Spectrom., 2001, 16, 652–657 RSC.
| This journal is © The Royal Society of Chemistry 2011 |