Graphene-polymer composite: extraction of polycyclic aromatic hydrocarbons from water samples by stir rod sorptive extraction†
Yan-Bo
Luo
a,
Jin-Sheng
Cheng
b,
Qiao
Ma
ac,
Yu-Qi
Feng
*a and
Jing-Hong
Li
*b
aKey Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), Department of Chemistry, Wuhan University, Wuhan, 430072, China. E-mail: yqfeng@whu.edu.cn; Fax: +86-27-68755595; Tel: +86-27-68755595
bDepartment of Chemistry, Key Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology, Tsinghua University, Beijing, 100084, China. E-mail: jhli@mail.tsinghua.edu.cn; Fax: +86-10-62771149; Tel: +86-10-62795270
cFocused Photonics (Hangzhou), Inc, 760 Bin,an Road, Binjiang District, Hangzhou, 310052, China
First published on 7th December 2010
Abstract
Due to the excellent mechanical, thermal and electrical properties, graphene/polymer composite is expected to have a variety of applications in analytical chemistry. In this study, a new poly(ethylene glycol dimethacrylate)/graphene composite was prepared by in situpolymerization. The new composite was used for the first time as the extraction coating of stir rod sorptive extraction for the preconcentration of polycyclic aromatic hydrocarbons (PAHs) from water samples. Because of the high specific surface area and π–π electrostatic stacking properties of graphene, the graphene-polymer composite showed higher extraction efficiencies towards most target PAHs from water samples than the neat polymer. Under the optimal conditions, a method for the determination of PAHs in water samples was proposed based on the combination of stir rod sorptive extraction (SRSE) and gas chromatography-mass spectrometry (GC-MS). The limit of detection (LODs) of the developed method for 16 PAHs ranged from 0.005 to 0.429 ng mL−1, depending on the compound. Good reproducibility of method was obtained as intra- and inter-day precisions, the relative standard deviations (RSDs) were less than 12.5% and 12.6%, respectively.
Introduction
Carbon materials, including graphite, carbon nanotubes (CNTs) and fullereneetc., have attracted great interdisciplinary interest due to their peculiar structural, mechanical and electrical properties.1 In past decades, carbon materials have been applied widely in analytical chemistry. For example, carbon nanotube-based electrodes have a wide range of electroanalytical applications2–6 because of their favorable properties of enhanced detection sensitivity and reduced fouling.7 With strong physical adsorption of organics,8carbon materials have been adopted as sorbents for the preconcentration of phenols,9–11 polychlorinated biphenyl,12 polybrominated diphenyl ethers13,14 and other organics15,16 in water samples. High adsorption capacity and complete reversible adsorption of polycyclic aromatic hydrocarbons (PAHs) by CNTs imply that CNTs are an excellent extraction adsorbent toward PAHs, however, the interaction between CNTs and PAHs is weak because of aggregation of CNTs in water.17As the mother of all graphitic forms,18graphene was experimentally produced in 2004.19 Due to its unique structure, thermal, mechanical and electrical properties,20,21graphene has been considered as a “rising-star” nanomaterial and thus has attracted much attention.22–24 Recently, graphene has been used in a wide range of materials with potential chemical, physical and engineering applications.25,26Graphene-based materials such as graphene-polymer composite27,28 are of interest due to the enhanced mechanical, thermal and electrical properties compared to neat polymer.27 Many strategies were adopted to prepare graphene-polymer composites. For example, thermoplastic graphene-polymer composites are usually prepared by melt intercalation technique. A thermoplastic polymer was mixed mechanically with graphene at elevated temperatures using injection molding.29 The polymer chains were then intercalated or exfoliated to form graphene-polymer composites. However, the extremely low bulk density of thermal reduction graphene sheets make it difficult to mix graphene into polymer matrix. Meanwhile, Stankovich et al. prepared graphene-polymer composite by a solution intercalation, which is based on a solvent system such as N,N-dimethylformamide (DMF) in which the polymer was dissolved and graphene layers were allowed to swell.27 The polymer then was adsorbed onto the graphene sheets. After the solvent is evaporated, graphene sheets reassemble and sandwiching the polymer to form the graphene-polymer composites. Although graphene has good dispersion in solvent, the solvent removal is a crucial issue in this method. More recently, Hu et al. reported a method for the preparation of graphene-polystyrene composite by in situpolymerization.30 In this method, graphene was first swollen within the monomer, and then polymerization was initiated either by heat or radiation after initiator was diffused. Although monomers have only been polymerized in solvents so far, a wide variety of graphene-polymer composites can be prepared using this method. The improvement of graphene-polymer composites in electrical properties makes it a suitable material for transparent conductors,31 supercapacitor electrodes,32 and sensor in the coming years for biomedical application. As advanced electrode materials, the potential applications of graphene-based material in analytical chemistry have been focused on electrochemical sensors, biosensors and electroanalysis.22,33–39 Significantly, as graphene has high specific surface area (theoretical calculated value of specific surface area, 2620 m2 g−1)34 and π–π electrostatic stacking property, graphene-polymer composite is expected to be a good sorbent material towards aromatic compounds. Therefore, it is of importance to exploit the potential applications of graphene as sorbent. To investigate the performance of graphene-polymer composites as sorbents, stir rod sorptive extraction (SRSE) was employed in our study. SRSE technique (Fig. 1S†) is an improved format of stir bar sorptive extraction (SBSE), which was proposed recently to avoid the friction loss of extraction coatings.40 Based on the SRSE device, a variety of materials may be utilized as extraction coatings to extract various analytes.
In this study, a poly(ethylene glycol dimethacrylate)-graphene composite was prepared by microwave irradiation method in a short time. DMF was used as porogenic solvent in the pre-polymerization mixture because graphene has good dispersion behavior in DMF.28 The new graphene-polymer composite was used as extraction coating of SRSE for PAHs extraction due to their strong π–π electrostatic interaction. For comparison, a neat polymer coating in the absence of graphene was also prepared in this study. Several parameters concerning extraction efficiency were investigated systematically. Under the optimal conditions, a PAHs detection method for water samples was proposed based on the combination of SRSE with gas chromatography-mass spectrometry (SRSE/GC-MS).
Experimental
Chemicals and materials
Ethylene glycol dimethacrylate (98%, EDMA) was obtained from Acros (New Jersey, USA). Polyethylene glycols 6000 (PEG 6000), diphenyl [Dip, internal standard (IS)], N,N-dimethylformamide (DMF) and methylene dichloride were supplied by Sinopharm Chemical Reagent (Shanghai, China). n-Hexane (95%, HPLC grade) and ethyl acetate (99.8%, HPLC grade) were supplied by CNW technologies GmbH (Dusseldorf, Germany). γ-Methacryloxypropyltrimethoxysilane (γ-MAPS) was obtained from the Chemical Plant of Wuhan University (Wuhan, China). Methanol (HPLC grade) was purchased from Concord Technology (Tianjin, China). Deionized water was purified using an Aike apparatus (Chengdu, China). The mixture containing 16 PAHs: naphthalene (NAPH), acenaphthylene (ACY), acenaphthene (ACP), fluorene (FLU), phenanthrene (PHEN), anthracene (ANTH), fluoranthene (FLT), pyrene (PYR), chrysene (CHRY), benzo[a]anthracene (BaA), benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF), benzo[a]pyrene (BaP), indeno[1,2,3-c,d]pyrene (INPY), dibenzo[a,h]anthracene (DiahA) and benzo[g,h,i]perylene (BghiP), at 0.2 mg mL−1 in n-hexane/methylene dichloride (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) was purchased from J&K Chemical Ltd. All working solutions of 16 PAHs were prepared in methanol and stored at 4 °C in the dark. Vial glass insert (31 × 6 mm, 0.1 mL) was supplied by Supelco (Pennsylvania, USA). Screw vial (2.0 mL) for desorption was purchased from Agilent (Palo Alto, USA).
:1, v/v) was purchased from J&K Chemical Ltd. All working solutions of 16 PAHs were prepared in methanol and stored at 4 °C in the dark. Vial glass insert (31 × 6 mm, 0.1 mL) was supplied by Supelco (Pennsylvania, USA). Screw vial (2.0 mL) for desorption was purchased from Agilent (Palo Alto, USA).
Preparation of graphene-polymer composite coated stir rod
Graphite oxide was synthesized according to the modified Hummers and Offeman method,41 and graphene was prepared by the reduction of graphene oxide using hydrazine as reductant (see ESI†). The preparation of graphene-polymer composite coating for SRSE was similar to the procedure described in our previous work with a minor modification.40 In brief, the pre-polymerization mixture was prepared by mixing EDMA (85.0 mg), DMF (270.0 μL), PEG 6000 (60.0 mg) and AIBN (1.0 mg) in a 1.5 mL polypropylene vial. After graphene was added to the above mixture, the solution was sonicated. The concentration of graphene in the pre-polymerization mixture was 1.0 mg mL−1. 150 μL of the pre-polymerization mixture was placed in an Eppendorf vial (0.6 mL). A vial glass insert was inserted into the Eppendorf vial and sealed with parafilm. After polymerization in microwave oven for 180 s, the vial glass insert with composite coating was carefully taken out of the Eppendorf vial, and washed with methanol water (1:1, v/v) and acetone in sequence. The neat polymer coated stir rod was prepared in the same way but without graphene. The composite and neat polymer coated stir rod were kept in deionized water before use.
Sample preparation
In this study, a lake- and tap-water sample were selected for investigation. The lake water sample was collected from East Lake, Wuhan city, and the tap water sample was taken from the laboratory after flowing for 10 min. Before the experiment, all water samples were filtered through 0.45 μm micropore membranes and stored in brown glass bottles at 4 °C in refrigerator.Instruments and analytical conditions
The powder X-ray diffraction (XRD) measurement of graphene was recorded on a Bruker D8-Advance X-ray powder diffractometer using Cu-Kα radiation (λ = 1.5406 Å) with scattering angles (2θ) of 8–60°. Raman spectroscopy (Renishaw microprobe RM1000, London, England) of graphene was recorded at a wavelength of 633 nm (He–Ne laser). An atomic force microscopic (AFM) image was taken using a Nanoscope III MultiMode SPM (Digital Instruments) with an AS-12 (“E”) scanner operated in tapping mode in conjunction with a V-shaped tapping tip (Applied Nanostructures SPM model: ACTA). The image was taken at a scan rate of 2 Hz. The morphology of the composite was examined using a scanning electron microscopy (SEM, Quanta 200, FEI, Holland). Nitrogen sorption experiments for the composite and neat polymer coating were carried out at −196 °C using JW–BK surface area and pore size analyzer (JWGB Sci. & Tech., Beijing, China). An autopore IV 9500 mercury porosimeter (Micromeritics Norcross, USA) was used to measure the through pore of the graphene-polymer composite coating. The microwave oven used in this work was a Galanz WP700 microwave oven (2450 MHz, Shunde, China) with a maximum power of 700 Watts.GC-MS analysis was performed using a Shimadzu GCMS-QP2010 Plus equipped with an AOC-20i+s autosampler (Kyoto, Japan). Data acquisition and analysis were performed using software from GCMS Solution (Shimadzu, Kyoto, Japan). The separation was achieved on a fused silica capillary column (Rxi®–5 ms, 30 m × 0.25 mm i.d., film thickness 0.25 μm) (Restek, Pennsylvania, USA). The oven temperature was programmed at 70 °C for 2.0 min, increased to 190 °C at a rate of 15 °C min−1 and held for 1.0 min, increased to 260 °C at a rate of 10 °C min−1 and finally to 285 °C at a rate of 5 °C min−1, where it was held for another 10.0 min. The solvent cut time was 4.5 min. The injection volume was 1.0 μL and splitless injection mode was used. The splitless time was 1.0 min. Helium (purity ≥ 99.999%) was used as the carrier gas at a flow rate of 1.20 mL min−1. The injection port, ion source and interface temperatures were 290, 220 and 280 °C, respectively. Selective ion monitoring mode was adopted for quantitative determination of the analytes. The MS program was set as follows: 4.5–8 min, m/z 128, 127, 129; 8–10.3 min, m/z 152, 153, 154, 151; 10.3–12 min, m/z 166, 165, 167; 12–14.5 min, m/z 178, 179, 176; 14.5–17.5 min, m/z 202, 203, 200; 17.5–21 min, m/z 228, 226, 229; 21–26 min, m/z 252, 253, 250; 26–30 min, m/z 276, 278, 277, 274.
Results and discussion
Preparation and characterization of graphene-polymer composite
Graphene has good dispersion behavior in polar solvents such as DMF.28,42 Hence, we prepared the new graphene-polymer composite on the basis of poly(ethylene glycol dimethacrylate), in which DMF was used as a porogenic solvent. Polymerization initiated by heating or UV light usually requires a long reaction time, and in such cases graphene in a pre-polymerization solution would be sedimentable during the polymerization process, leading to heterogeneous dispersion of graphene in polymer. As an alternative method, microwave irradiation has been proposed. Despite the fact that the mechanism of microwave irradiation on chemical reactions is still under debate,43 microwave irradiation can significantly increase the reaction rate. Furthermore, microwave irradiation was reported to have less effect on the physical properties of polymers.44 Thus, microwave irradiation was adopted for the preparation of graphene-polymer composite. Optimization of microwave irradiation power (259, 280, 462, 595 and 700 W) for the polymerization was carried out. With increasing microwave irradiation, less reaction time was required to complete polymerization. To effectively avoid sedimentation of graphene during polymerization, 700 W of microwave irradiation power was chosen for the preparation of graphene-polymer composite, and the reaction time was set at 180 s.In Fig. 1a the AFM image shows that the graphene nanosheets were mostly single layer (height ∼0.8 nm). XRD patterns in Fig. 1b reveals that the graphene nanosheets’ peak at 2θ = 24.7°, the characteristic peak of graphene.23Raman spectrum of graphene in Fig. 1c displays two prominent peaks at 1587 and 1332 cm−1, which correspond to the well-documented G and D bands, respectively.23
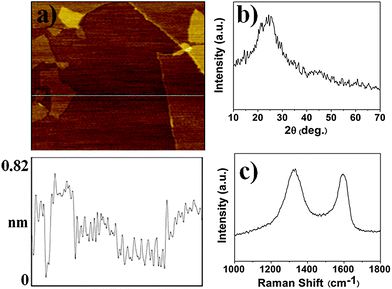 | ||
| Fig. 1 AFM image and depth profile on (a) mica substrate (size 2.5 × 2.5 μm), (b) XRD pattern and (c) Raman spectroscopy of graphene. | ||
The total specific surface area of the neat polymer and graphene-polymer composite materials was determined by N2 sorption method. The Barrett–Joyner–Halenda (BJH) plot average pore diameters of the two materials were found to be 5.1 and 4.9 nm, respectively; the total specific surface area of the two materials were 34 and 149 m2 g−1 through Brunauer–Emmett–Teller (BET) plot. Compared with neat polymer in the absence of graphene, graphene-polymer composite had high surface area that may ensure its higher extraction capacity towards target analytes.
The through pore size distribution of graphene-polymer composite was determined by mercury porosimeter (Fig. 2). The though pore size was found to be around 1.4 μm and the pore size distribution was narrow, which would be favorable for mass transfer during extraction applications. The morphology of graphene-polymer composite was examined by SEM, and a typical micrography is displayed in Fig. 2. The interconnected skeletons and interconnected textural pore of the materials could be clearly observed. It could be seen from Fig. 2 that the graphene was dispersed and embedded throughout the polymer matrix and an interconnected graphene network was formed.
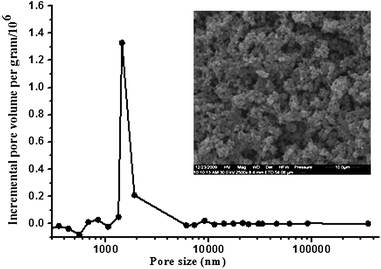 | ||
| Fig. 2 Incremental pore size distribution profiles of graphene-polymer composite measured by mercury porosimeter. The inset shows scanning electron microscope image of graphene-polymer composite with a magnification 2500. | ||
Raman spectra of neat polymer and graphene-polymer composite were recorded at a wavelength of 633 nm (He–Ne laser) by LabRAM HR 800 UV Laser Confocal Raman Microspectroscopy. As shown in Fig. 3, two prominent peaks at 1588 and 1325 cm−1 became obvious by increasing the concentration of graphene in pre-polymerization mixture, which correspond to the Raman shift of the G and D bands of graphene. The Raman spectra also demonstrated that graphene was successfully incorporated into the matrix.
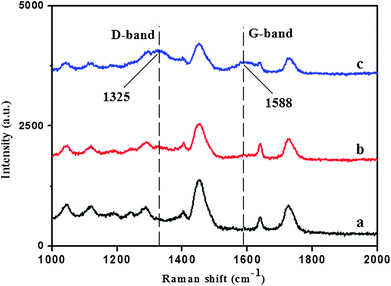 | ||
| Fig. 3 Raman spectra of (a) neat polymer and (b, c) graphene-polymer composite. The concentrations of graphene in pre-polymerization mixtures were 0.5 mg mL−1 (b) and 1.0 mg mL−1 (c). | ||
Optimization of the extraction parameters
Considering the high specific surface area and π–π electrostatic stacking property of graphene, graphene-polymer composite was prepared and used as a new extraction coating of SRSE. In order to achieve the best extraction efficiency of the new coating for 16 PAHs, several parameters, including desorption solvents, salt concentration, organic phase content and extraction time, were investigated.:1, v/v) were used as desorption solvents to determine the impact on the recoveries. It was found that n-hexane is the best desorption solvent (see ESI†).
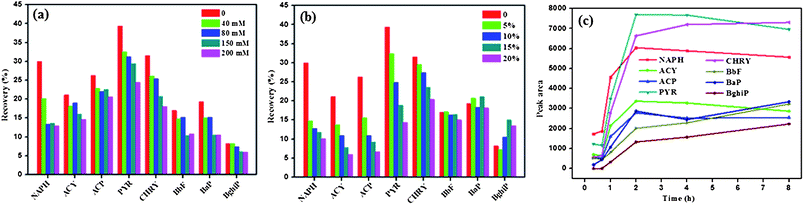 | ||
| Fig. 4 (a) Effect of content of inorganic salt in the sample solution on extraction efficiency. Sample solutions with 16 PAHs spiked at 1 ng mL−1 were prepared with different concentrations of NaCl, the extraction time was 90 min, no additional organic phase was used. (b) Effect of content of organic phase in the sample solution on extraction efficiency. Sample solutions with different concentration of methanol containing 16 PAHs at 1 ng mL−1 were used for the extraction, the extraction time was 90 min, no additional inorganic salt was used. (c) Equilibrium extraction time profiles of representative PAHs for SRSE. Sample solutions with 16 PAHs spiked at 1 ng mL−1 were extracted from 20 to 240 min, no additional inorganic salt and organic phase were used. Recovery = Cd/Cs × 100%, where Cd and Cs are the peak areas of 16 PAHs of desorption solution and standard solution (50 ng mL−1) obtained by GC-MS analysis, respectively. | ||
On the basis of the above discussion, the optimal conditions for SRSE are concluded as follows: 120 min for extraction; the stir rod (glass insert with composite coating) was immersed in 1.0 mL n-hexane in a screw vial and shaken at 40 °C for 20 min for desorption; no salt and organic phase were added to the sample solution.
Comparison with neat polymer
Under optimal conditions, both the graphene-polymer composite coating and neat polymer coating were applied to extract 16 PAHs from a spiked water sample of 5 ng mL−1via SRSE. The obtained peak areas of 16 PAHs are shown in Fig. 5. The peak area ratios of the extracted PAHs with graphene-polymer composite coating compared to those with neat polymer coating were in the range from 0.91 to 1.47 for 16 PAHs. This result indicates that graphene-polymer composite coating exhibited higher extraction efficiencies towards most target analytes than did the neat polymer coating. This is possibly due to carbon atoms in the plane of graphene possessing mixed hybridization of sp2 and sp3; therefore, there is a highly delocalized conjugate system of the π-electron, which enhances the π–π interaction with the aromatic PAHs. On the other hand, graphene has a large specific surface area. Furthermore, a large sorption surface on the planar surface of graphene also has strong binding affinity for planar molecules. All these indicate that the graphene-polymer composite has higher extraction efficiency for PAHs. In our experiments, no cracking of the graphene-polymer composite coating was observed during stirring, which indicates that the mechanical stability of the coating was good. Meanwhile, in the extraction process the composite coating also showed good rod to rod repeatability; the intra-day RSDs was less than 12.5% (three different stir rods).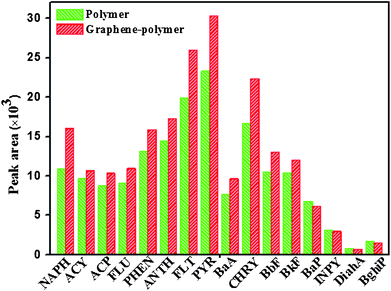 | ||
| Fig. 5 Peak areas of 16 PAHs extracted with graphene-polymer composite and neat polymer coating under the optimized conditions. Conditions: no salt and organic phase were used; extraction time, 120 min; desorption time, 20 min; desorption solvent, 1.0 mL n-hexane. | ||
Validation of the SRSE/GC-MS method
In the calibration of this method, variable amounts of stock PAHs mixtures and a fixed volume (100 μL of 2.5 μg mL−1 Dip) of IS solution were added to deionized water to prepare the spiked working standard solutions which were then extracted by SRSE. The linear regression analyses were performed using peak areas ratios (PAHs/IS) against the respective analytes concentration. Limit of detection (LODs) and limit of quantitation (LOQs) were calculated as the concentration corresponding to a signal 3 and 10 times the standard deviation of the baseline noise, respectively. The calibration curves, LODs and LOQs data are listed in Table 1. The LODs and LOQs for 16 PAHs were found to be 0.005–0.429 ng mL−1 and 0.016–1.429 ng mL−1, respectively. The range of LODs and LOQs values were wide, depending on the both overall recoveries and MS intensities of the studied PAHs. Correlation coefficients (R) between 0.9811 and 0.9989 were obtained, showing a high degree of correlation between concentration and peak area ratios.| Analytes | Concentration range/ng mL−1 | Regression line | LOD/ng mL−1 | LOQ/ng mL−1 | ||
|---|---|---|---|---|---|---|
| Slope | Intercept | R2 | ||||
| Naphthalene | 0.1-200 | 0.0670 | −0.1653 | 0.9972 | 0.026 | 0.087 |
| Acenaphthylene | 0.1-200 | 0.0689 | −0.1580 | 0.9968 | 0.019 | 0.062 |
| Acenaphthene | 0.1-200 | 0.0541 | −0.0963 | 0.9979 | 0.013 | 0.044 |
| Fluorene | 0.2-200 | 0.0611 | −0.1503 | 0.9968 | 0.034 | 0.112 |
| Phenanthrene | 0.1-200 | 0.1019 | −0.1600 | 0.9973 | 0.015 | 0.048 |
| Anthracene | 0.1-200 | 0.0765 | −0.0521 | 0.9977 | 0.019 | 0.063 |
| Fluoranthene | 0.1-200 | 0.1339 | −0.1510 | 0.9966 | 0.006 | 0.021 |
| Pyrene | 0.1-200 | 0.1514 | −0.1557 | 0.9978 | 0.005 | 0.016 |
| Benzo[a]anthracene | 0.1-200 | 0.0240 | −0.0676 | 0.9894 | 0.019 | 0.064 |
| Chrysene | 0.1-200 | 0.0276 | −0.0272 | 0.9941 | 0.010 | 0.034 |
| Benzo[b]fluoranthene | 0.1-200 | 0.0219 | −0.0087 | 0.9880 | 0.022 | 0.074 |
| Benzo[k]fluoranthene | 0.1-200 | 0.0293 | −0.0417 | 0.9862 | 0.018 | 0.060 |
| Benzo[a]pyrene | 0.2-200 | 0.0252 | −0.0613 | 0.9889 | 0.056 | 0.185 |
| Indeno[1,2,3-c,d]pyrene | 1-200 | 0.0259 | −0.2110 | 0.9626 | 0.167 | 0.556 |
| Dibenzo[a,h]anthracene | 5-200 | 0.0197 | −0.2401 | 0.9685 | 0.429 | 1.43 |
| Benzo[g,h,i]perylene | 1-200 | 0.0266 | −0.1334 | 0.9823 | 0.130 | 0.435 |
In this study, the precision of the method was investigated by determining intra- and inter-day relative standard deviations (RSDs) of the analyses. The intra- and inter-day RSDs were calculated with 16 PAHs spiked at three different concentrations in water. The RSDs data in water samples are summarized in Table 2. The intra- and inter-day RSDs of 16 PAHs were less than 12.5% and 12.6%, respectively, which should be satisfactory for determining the PAHs in water matrix.
| Analytes | Intra-day precision (RSDs, n = 3) | Inter-day precision (RSDs, n = 3) | ||||
|---|---|---|---|---|---|---|
| 1 ng mL−1 | 10 ng mL−1 | 100 ng mL−1 | 1 ng mL−1 | 10 ng mL−1 | 100 ng mL−1 | |
| a nd, no data. | ||||||
| Naphthalene | 9.8 | 6.4 | 5.2 | 8.4 | 9.2 | 10.9 |
| Acenaphthylene | 1.7 | 9.0 | 6.3 | 2.0 | 5.3 | 5.4 |
| Acenaphthene | 9.9 | 4.8 | 4.1 | 1.2 | 1.7 | 6.0 |
| Fluorene | 2.5 | 7.1 | 6.5 | 4.1 | 3.0 | 4.8 |
| Phenanthrene | 1.2 | 4.8 | 9.4 | 2.1 | 7.5 | 1.6 |
| Anthracene | 5.2 | 10.7 | 4.2 | 4.8 | 6.3 | 4.2 |
| Fluoranthene | 6.0 | 8.0 | 1.5 | 2.9 | 12.3 | 6.1 |
| Pyrene | 6.7 | 6.7 | 1.4 | 1.6 | 10.8 | 7.0 |
| Benzo[a]anthracene | 6.0 | 11.1 | 9.9 | 5.3 | 0.2 | 1.0 |
| Chrysene | 3.7 | 9.8 | 12.5 | 12.6 | 6.2 | 4.8 |
| Benzo[b]fluoranthene | 4.5 | 1.2 | 3.3 | 12.3 | 9.1 | 1.3 |
| Benzo[k]fluoranthene | 6.7 | 7.3 | 2.7 | 4.6 | 6.2 | 2.3 |
| Benzo[a]pyrene | 3.9 | 6.5 | 10.8 | 8.1 | 5.6 | 1.2 |
| Indeno[1,2,3-c,d]pyrene | 3.7 | 6.5 | 4.7 | 5.5 | 10.9 | 5.0 |
| Dibenzo[a,h]anthracene | nd a | 6.8 | 4.4 | nd | 9.1 | 12.0 |
| Benzo[g,h,i]perylene | 4.1 | 9.3 | 3.4 | 7.0 | 11.2 | 9.0 |
Application to real sample
Under the optimized conditions, the developed method was successfully applied to the analysis of trace PAHs in real water samples. The recoveries were determined by comparing the calculated amounts PAHs from the spiked real samples with the total spiking amounts (10 ng mL−1). The precision was estimated by performing three extractions at the spiked concentration of 10 ng mL−1 for real samples. The results are given in Table 3. Wu et al. reported the existence of anthracene and pyrene in the surface water of East Lake.46 Our results confirmed their existence; additionally fluoranthene was detected as well. The concentrations were 0.71, 1.04 and 1.14 ng mL−1, respectively.| Analytes | Tap water | Lake water | ||||
|---|---|---|---|---|---|---|
| Concentration/ng mL−1 | Found/ng mL−1 | Recovery (%±RSDs, n = 3) | Concentration/ng mL−1 | Found/ng mL−1 | Recovery (%±RSDs, n = 3) | |
| a Tap water and lake water were spiked at 10 ng mL−1. b nd, not detected. | ||||||
| Naphthalene | nd b | 8.08 | 80.8 ± 3.6 | nd | 8.43 | 84.3 ± 3.7 |
| Acenaphthylene | nd | 8.71 | 87.1 ± 6.6 | nd | 8.91 | 89.1 ± 2.7 |
| Acenaphthene | nd | 9.24 | 92.4 ± 3.2 | nd | 8.94 | 89.4 ± 4.7 |
| Fluorene | nd | 8.96 | 89.6 ± 1.4 | nd | 8.55 | 85.5 ± 7.3 |
| Phenanthrene | nd | 7.10 | 71.0 ± 8.3 | nd | 7.02 | 70.2 ± 11.3 |
| Anthracene | nd | 8.19 | 81.9 ± 9.6 | 0.71 | 8.66 | 79.5 ± 10.2 |
| Fluoranthene | nd | 6.98 | 69.8 ± 1.4 | 1.14 | 6.67 | 55.3 ± 14.7 |
| Pyrene | nd | 6.72 | 67.2 ± 3.7 | 1.04 | 6.49 | 54.5 ± 15.5 |
| Benzo[a]anthracene | nd | 9.21 | 92.1 ± 14.6 | nd | 9.88 | 98.8 ± 18.4 |
| Chrysene | nd | 10.95 | 109.5 ± 8.0 | nd | 10.60 | 106.0 ± 4.9 |
| Benzo[b]fluoranthene | nd | 7.93 | 79.3 ± 3.8 | nd | 7.52 | 75.2 ± 16.8 |
| Benzo[k]fluoranthene | nd | 7.16 | 71.6 ± 14.6 | nd | 7.34 | 73.4 ± 7.4 |
| Benzo[a]pyrene | nd | 6.34 | 63.4 ± 8.7 | nd | 6.29 | 62.9 ± 11.4 |
| Indeno[1,2,3-c,d]pyrene | nd | 11.08 | 110.8 ± 4.1 | nd | 10.80 | 108.0 ± 7.4 |
| Dibenzo[a,h]anthracene | nd | 14.44 | 144.4 ± 1.7 | nd | 14.19 | 141.9 ± 4.7 |
| Benzo[g,h,i]perylene | nd | 8.13 | 81.3 ± 3.6 | nd | 7.61 | 76.1 ± 10.0 |
Conclusions
In this work, a new poly(ethylene glycol dimethacrylate)-graphene composite coated stir rod sorptive extraction was prepared and applied for the preconcentration of PAHs from water samples. Compared with the neat polymer, due to the excellent π–π electrostatic stacking property of graphene and high surface specific area, the graphene-polymer composite showed higher extraction efficiencies for most target PAHs from water samples. Under optimal conditions, a method was proposed based on the combination of SRSE and gas chromatography-mass spectrometry (SRSE/GC-MS), and applied to determinate PAHs in the environment from water samples.Acknowledgements
This work is partly supported by grants from the National Science Fund for Distinguished Young Scholars (No. 20625516), the Science Fund for Creative Research Groups (No. 20921062), NSFC and National Key Technologies R&D Program (2006BAF07B03 and 2007CB310500).References
- L. M. Ravelo-Perez, M. Asensio-Ramos, J. Hernandez-Borges and M. A. Rodriguez-Delgado, Electrophoresis, 2009, 30, 1624–1646 CrossRef CAS.
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Anal. Chim. Acta, 2008, 618, 54–60 CrossRef CAS.
- C. B. Jacobs, M. J. Peairs and B. J. Venton, Anal. Chim. Acta, 2010, 662, 105–127 CrossRef CAS.
- K. B. Wu, S. S. Hu, J. J. Fei and W. Bai, Anal. Chim. Acta, 2003, 489, 215–221 CrossRef CAS.
- P. H. Yang, W. Z. Wei and C. Y. Tao, Anal. Chim. Acta, 2007, 585, 331–336 CrossRef CAS.
- N. S. Lawrence, R. P. Deo and J. Wang, Anal. Chim. Acta, 2004, 517, 131–137 CrossRef CAS.
- I. Dumitrescu, P. R. Unwin and J. V. Macpherson, Chem. Commun., 2009, 45, 6886–6901 Search PubMed.
- C. Saridara, R. Brukh, Z. Iqbal and S. Mitra, Anal. Chem., 2005, 77, 1183–1187 CrossRef CAS.
- X. Y. Liu, Y. S. Ji, Y. H. Zhang, H. X. Zhang and M. C. Liu, J. Chromatogr., A, 2007, 1165, 10–17 CrossRef CAS.
- Q. L. Li, X. F. Wang and D. X. Yuan, J. Chromatogr., A, 2009, 1216, 1305–1311 CrossRef CAS.
- S. Y. Zhu, W. X. Niu, H. J. Li, S. Han and G. B. Xu, Talanta, 2009, 79, 1441–1445 CrossRef CAS.
- J. X. Yu, L. Dong, C. Y. Wu, L. Wu and J. Xing, J. Chromatogr., A, 2002, 978, 37–48 CrossRef CAS.
- W. Y. Zhang, Y. Sun, C. Y. Wu, J. Xing and J. Y. Li, Anal. Chem., 2009, 81, 2912–2920 CrossRef CAS.
- J. X. Wang, D. Q. Jiang, Z. Y. Gu and X. P. Yan, J. Chromatogr., A, 2006, 1137, 8–14 CrossRef CAS.
- Q. L. Li, X. X. Ma, D. X. Yuan and J. S. Chen, J. Chromatogr., A, 2010, 1217, 2191–2196 CrossRef CAS.
- H. Y. Niu, Y. Q. Cai, Y. L. Shi, F. S. Wei, J. M. Liu, S. F. Mou and G. B. Jiang, Anal. Chim. Acta, 2007, 594, 81–92 CrossRef CAS.
- K. Yang and B. S. Xing, Environ. Pollut., 2007, 145, 529–537 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- H. X. Chang, L. H. Tang, Y. Wang, J. H. Jiang and J. H. Li, Anal. Chem., 2010, 82, 2341–2346 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- Y. Wang, Y. M. Li, L. H. Tang, J. Lu and J. H. Li, Electrochem. Commun., 2009, 11, 889–892 CrossRef CAS.
- C. N. R. Rao, K. Biswas, K. S. Subrahmanyam and A. Govindaraj, J. Mater. Chem., 2009, 19, 2457–2469 RSC.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- W. Choi, I. Lahiri, R. Seelaboyina and Y. S. Kang, Crit. Rev. Solid State Mat. Sci., 2010, 35, 52–71 Search PubMed.
- C. Soldano, A. Mahmood and E. Dujardin, Carbon, 2010, 48, 2127–2150 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS.
- K. Kalaitzidou, H. Fukushima and L. T. Drzal, Carbon, 2007, 45, 1446–1452 CrossRef CAS.
- H. T. Hu, X. B. Wang, J. C. Wang, L. Wan, F. M. Liu, H. Zheng, R. Chen and C. H. Xu, Chem. Phys. Lett., 2010, 484, 247–253 CrossRef CAS.
- S. Watcharotone, D. A. Dikin, S. Stankovich, R. Piner, I. Jung, G. H. B. Dommett, G. Evmenenko, S. E. Wu, S. F. Chen, C. P. Liu, S. T. Nguyen and R. S. Ruoff, Nano Lett., 2007, 7, 1888–1892 CrossRef CAS.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. S. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- X. Chen, C. L. Fu and W. S. Yang, Analyst, 2009, 134, 2135–2140 RSC.
- Y. M. Li, L. H. Tang and J. H. Li, Electrochem. Commun., 2009, 11, 846–849 CrossRef CAS.
- J. Li, S. J. Guo, Y. M. Zhai and E. K. Wang, Electrochem. Commun., 2009, 11, 1085–1088 CrossRef CAS.
- J. F. Wu, M. Q. Xu and G. C. Zhao, Electrochem. Commun., 2010, 12, 175–177 CrossRef CAS.
- H. F. Xu, H. Dai and G. N. Chen, Talanta, 2010, 81, 334–338 CrossRef CAS.
- J. Li, S. J. Guo, Y. M. Zhai and E. K. Wang, Anal. Chim. Acta, 2009, 649, 196–201 CrossRef CAS.
- K. P. Liu, J. J. Zhang, G. H. Yang, C. M. Wang and J. J. Zhu, Electrochem. Commun., 2010, 12, 402–405 CrossRef CAS.
- Y. B. Luo, Q. Ma and Y. Q. Feng, J. Chromatogr., A, 2010, 1217, 3583–3589 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS.
- D. Bogdal, P. Penczek, J. Pielichowski and A. Prociak, Adv. Polym. Sci., 2003, 163, 193–263 CAS.
- J. A. Li, X. L. Zhu, J. Zhu and Z. P. Cheng, Radiat. Phys. Chem., 2007, 76, 23–26 CrossRef CAS.
- V. G. Zuin, L. Montero, C. Bauer and P. Popp, J. Chromatogr., A, 2005, 1091, 2–10 CrossRef.
- Y. L. Wu, L. B. Xia, R. Chen and B. Hu, Talanta, 2008, 74, 470–477 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of graphite oxide and graphene. See DOI: 10.1039/c0ay00624f |
| This journal is © The Royal Society of Chemistry 2011 |