Quantification of ellagic acid in cosmetic products by using a partially preanodized screen-printed carbon electrode coupled with flow injection analysis
Pei-Yen
Chen
a,
Ying-Ming
Ji
a,
Chin-Hsiang
Luo
b,
Yi-Shyan
Chen
a and
Ying
Shih
*a
aDepartment of Cosmetic Science Providence University, 200 Chungchi Rd., Taichung, 43301, Taiwan. E-mail: yingshih@pu.edu.tw; Fax: +886-4-26311167; Tel: +886-4-26328001
bDepartment of Safety, Health and Environmental Engineering, 34 Chungchi Rd., Taichung, 43302, Taiwan
First published on 26th November 2010
Abstract
A partially preanodized screen-printed carbon electrode (PSPCE*) coupled with flow injection analysis (FIA) was developed to raise the selectivity of ellagic acid (EA). To confirm the effectiveness of partial preanodization, two pretreated screen-printed carbon electrodes were electrochemically compared. One was a PSPCE* fabricated by potential cycling (−1.0 – +1.0 V vs.Ag/AgCl) to make the electrode surface partially preanodized and the other was a SPCE* fabricated by a high treatment potential (+2.0 V vs.Ag/AgCl). Cyclic voltammograms showed that the catalytic current of EA was observed at both the PSPCE* and SPCE*. No catalytic current of ascorbic acid 2-glucoside (AA2G) and magnesium ascorbyl phosphate (MAP) was observed at the PSPCE*. The PSPCE* selectively detected EA. The factors, which influence the EA response current, have previously been discussed. At the detection limit (0.012 ppm, S/N = 3), the linear calibration plot (R2 = 0.998) was attained for 0.1–50 ppm of EA solutions. A relative standard deviation of 4.37 and 3.90% was conducted for consecutive injections (n = 10) of 1 and 50 ppm EA, respectively. Finally, a practical application of the proposed method was demonstrated by the quantitative analysis of EA in skin whitening creams, and good recovery was obtained.
1. Introduction
Ellagic acid (EA) is one of a number of polyphenolic compounds found in berries and pomegranates.1 Studies have confirmed the anti-mutagenic, antiviral, and anti-oxidative properties of EA.2–4EA is a popular ingredient in cosmetic products because of its whitening effect on skin.5 The level of EA additive is limited to 0.5% (w/w) for commercial cosmetics in Taiwan. Several chromatographic techniques have been proposed to detect the amount of EA in fruit juices, muscadine grapes and sheep ruminal fluid.6–12 Few works, however, addressed the EA levels in cosmetic products.During preanodization, the screen-printed carbon electrode (SPCE) tends to become more porous because its surface reorientation produces surface carbonyl functionalities. Preanodized(*) SPCE, SPCE*, has been used to detect several analytes of improved electrochemical activity and lower over potential in voltammetry.13–17SPCE* treated with oxygen plasma was used to identify dopamine, uric acid and ascorbic acid because a substantial increase of carbonyl functional groups appeared on the electrode surfaces.18 The pretreated glassy carbon electrode is used to analyze hydroquinone which has a structure similar to EA.19 However, the SPCE* must be used with an amperometric separation technique to enhance its poor selectivity of analytes with similar electrochemical properties.20,21 Therefore, it is necessary for cosmetic EA detection which has advantages of simplicity, short analysis time, low cost, high sensitivity, selectivity and reproducibility.
In this work, two electrochemically pretreated SPCEs (SPCE* and PSPCE*) were compared for their ability to identify EA and selectively detect EA from the co-existing species, ascorbic acid 2-glucoside (AA2G) and magnesium ascorbyl phosphate (MAP). For fast detection of EA, the PSPCE* was combined with flow injection analysis (FIA). The factors that influence the response current in FIA such as detection potential and flow rate were investigated. A practical application of this investigation was demonstrated by performing selective measurements of EA in commercially available creams for skin whitening.
2. Experimental
2.1 Materials and instruments
EA, AA2G and MAP (Sigma-Aldrich, USA) and sodium hydroxide (Showa, Japan) were used. All reagents were of analytical reagent grade and prepared with de-ionized (D.I.) water. Cosmetic cream samples were purchased from local supermarkets. Cyclic voltammetric (CV), and amperometric experiments were performed using a CHI800c electrochemical workstation (USA). Scanning electron microscopy (SEM) (Topcon ABT-150S, Japan) and energy-dispersive X-ray spectroscopy (EDX) and Electron Spectroscopy for Chemical Analysis (ESCA)(ULVAC-PHI,PHI5000, ST Instrument, Netherlands) were used to detect the morphological and elemental properties of the PSPCE* and SPCE*. A SPCE with a geometric area of 0.196 cm2 (SE100) and a wall-jet flow cell were purchased from Zensor R&D (Taiwan).2.2 Electrode preparation
The SPCE* was pretreated according our prior work:13 (1) scanning the potential in a range of 0.0 to 1.40 V (vs.Ag/AgCl) for cyclic voltammetry in a phosphate buffer solution (0.1 mol L−1, pH 7) and; (2) applying 2.0 V, 240 s to the electrode in the 0.1 mol L−1NaOH using amperometry. The PSPCE* was pretreated by scanning the potential in ranges of −1.0 up to +1.0 V (vs.Ag/AgCl) in 0.1 mol L−1NaOH by CV of 20 cycles.2.3 Flow injection analysis
Flow injection analysis (FIA) was performed by a Rheodyne 7125 sample injection valve (20 μL loop) and equilibrated in 0.025 mol L−1NaOH carrier solution at +0.40V vs.Ag/AgCl until the current became constant, which took approximately 5 min at room temperature. The EA oxidation peak signal was uniformly taken as a quantitative parameter.2.4 Preparation of sample solutions
Test solutions of two commercial samples were prepared by dissolving 0.2000 g (sample 1) and 0.1000 g (sample 2) of the skin whitening cream (0.5% (w/w) EA containing) in 100 mL of supporting electrolyte. Next, the solution was filtered through a 0.22 μm filter paper to remove the suspending colloids. Finally, the filtered solution was diluted to the concentration required for the FIA.3. Results and discussion
3.1 Cyclic voltammetry
Fig.1 illustrates the CV response of EA (100 ppm) with the corresponding background voltammogram at a bare screen-printed carbon electrode (SPCE), PSPCE* and SPCE* in 0.1 mol L−1NaOH. No oxidation peak was observed for the SPCE (Fig. 1A). For the PSPCE*, a peak was observed around 0.0 V (vs.Ag/AgCl) and the current level increased (Fig. 1B). In Fig. 1C, two oxidation peaks were observed for the SPCE* at potentials of 0.0 and 0.25 V. Therefore, both the PSPCE* and SPCE* showed the catalytic current of EA. | ||
Fig. 1 CV responses for (A) the SPCE, (B) PSPCE*, and (C) SPCE* in 0.1 mol L−1NaOH in the absence (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and presence ( ) and presence (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) of 100 ppm EA at a scan rate of 50 mV s−1. ) of 100 ppm EA at a scan rate of 50 mV s−1. | ||
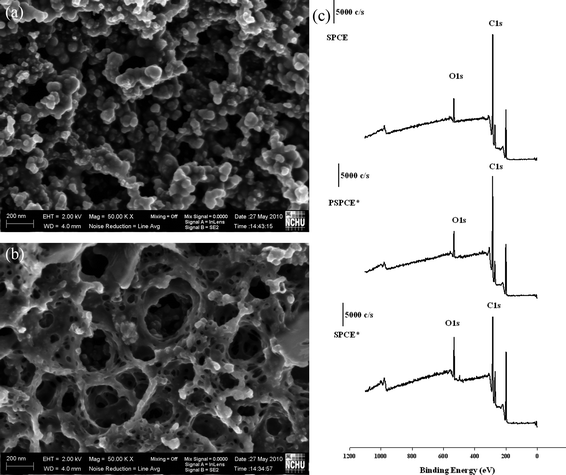
Two ascorbic acid derivatives, AA2G and MAP, were used to evaluate the selectivity and ingredient interference on the PSPCE* and SPCE*. Fig. 2A and 2B show that no obvious current occurred on the PSPCE* when 50 ppm AA2G and MAP were added. However, two clear oxidation peaks were observed around +0.33 and +0.69 V for the SPCE* (Fig. 2C and 2D). The PSPCE* produced a lower catalytic current than the SPCE*, but its selectivity was better when using amperometry. SEM and ESCA was used to detect the morphological and surface properties of the PSPCE* and SPCE*. Fig. 3 shows the results. The PSPCE* is less porous than the SPCE*. According to the ESCA results (Fig. 3C), the ratio of O1s/C1s (0.215) of the SPCE, 0.241 of the PSPCE* and 0.405 of the SPCE*. The SPCE* shows higher O1s/C1s ratio than the PSPCE*. Since the oxygen level can influence the electrocatalytic properties,17 the sensitivity of PSPCE* for EA is less than that of SPCE*. The PSPCE* showed a higher selectivity for EA than the SPCE*.
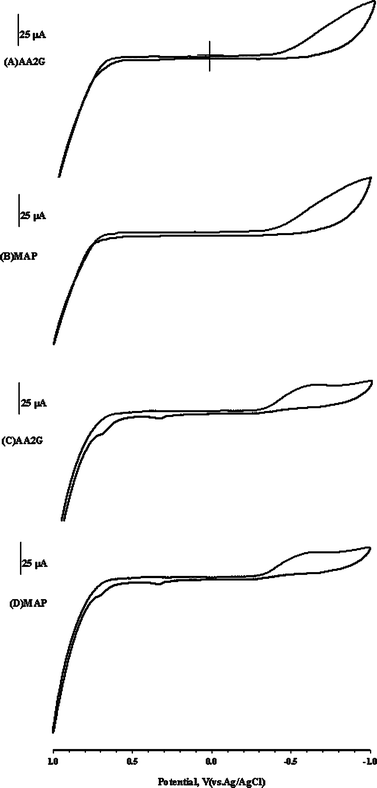 | ||
| Fig. 2 CV response of 50 ppm AA2G (A,C) and MAP (B,D) for PSPCE* and SPCE*, respectively, with 0.1 mol L−1NaOH at a scan rate of 50 mV s−1. | ||
In Fig.4, the PSPCE* was prepared in various NaOH concentrations and the catalytic current measured at 50 ppm of EA. How the increase with an increase of NaOH concentration during PSPCE* preparation is displayed. When the concentration of NaOH was larger than 0.1 mol L−1, the strong base destroyed the isolating layer on the SPCE surfaces. Thus, 0.1 mol L−1 of NaOH was used in electrode preparation.
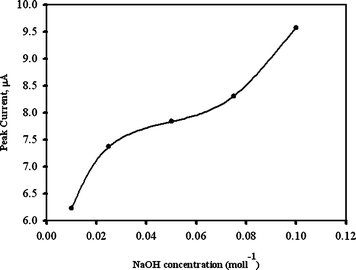 | ||
| Fig. 4 Typical plot of various concentration of NaOHversus CV peak current in electrode preparation with 50 ppm of EA added. | ||
3.2 Flow injection analysis
A wall-jet type flow cell was used in this study. The FIA parameters were optimized for the purpose of analytical application. The optimization of applied potential, flow rate and carrier solution in the detection of EA was studied. Fig. 5 shows that typical FIA response plots for 50 ppm of EA under various applied potentials (A), flow rates (B) and various NaOH concentration (C). In Fig. 5A, the applied potential variation and the FIA peak currents were increased in the potential window of −0.05 to +0.5 V (vs.Ag/AgCl). It is known that a potential over 0.4 V for other electrochemically active compounds will also be detected. Thus, 0.4 V was used for the detection potential. In Fig. 5B, the flow rate ranged from 0.1 to 0.5 mL min−1 and were employed to detect the FIA peak currents in 0.025 mol L−1NaOH. The tendency of the peak current was to plateau after 0.3 mL min−1. In Fig. 5C, various NaOH concentrations were studied from 0.025 mol L-1 to 0.12 mol L-1 and no obvious difference was observed. Therefore, a flow rate of 0.3 mL min−1, an applied potential of +0.4 V and 0.025 mol L−1NaOH were chosen as optimal values for further experiments. Fig. 6A shows the FIA peak current responses for increasing EA concentrations under these optimized conditions. A linear regression coefficient of 0.998 for detecting EA of 0.1–50 ppm was obtained (with linear equation Y = 0.0177X + 0.0218). A detection limit of 0.012 ppm was obtained for a signal-to-noise ratio of 3. This was superior to previous chromatographic techniques.8–12Fig. 6C and 6D shows the actual FIA graph of the selectivity including kojic acid (KA), arbutin (AR), AA2G, MAP and extract of green tea (Gr) under detection potentials of 0.4 V and 0.5 V. The AR and Gr showed the oxidation signal in both 0.4 V and 0.5 V. However, the EA signal obviously decreases at 0.5 V, this indicates that the electrode is more stable at 0.4 V. It is very hard to find EA, AR and Gr coexisting in the skin whitening cream, but our further study indicated that the influence of AR and Gr can be overcome, when the detection potential of 0.3 V is applied (data not enclosed).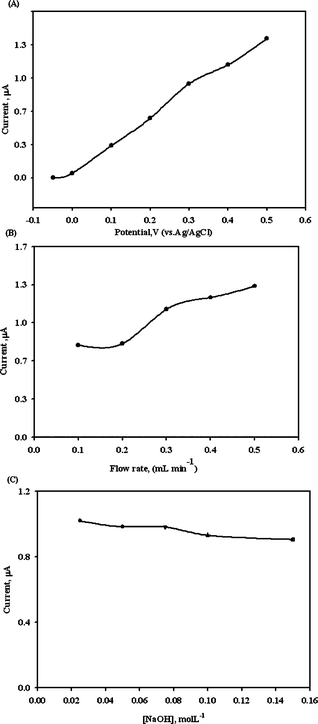 | ||
| Fig. 5 Effects of applied potential (A), flow rate (B) and carrier solution (C) on the detection of 50 ppm EA for PSPCE*. | ||
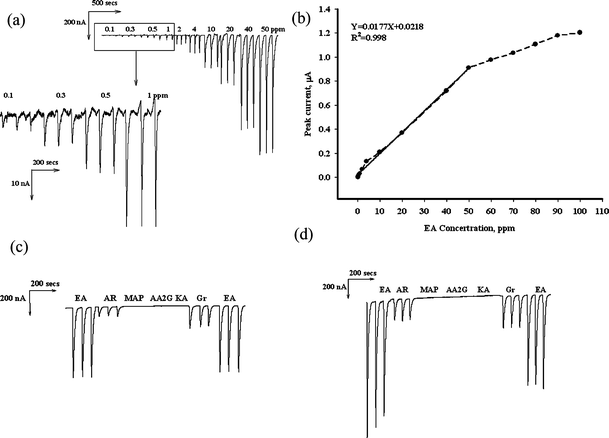 | ||
| Fig. 6 (A) FIA responses of PSPCE* with increasing concentration of EA in 0.025 mol L−1NaOH carrier solution. (B) typical plot of current versus various EA concentration. Selectivity study within 50 ppm EA, AR, MAP, AA2G, KA and Gr at detection potential 0.4 V (C) and 0.5 V (D). Optimized conditions: Applied potential of +0.40 V vs.Ag/AgCl and flow rate of 0.3 mL min−1 in 0.025 mol L-1NaOH. | ||
Duplicate measurements were used to analyze EA concentrations of 1 and 50 ppm for continuous injections (n = 10). The relative standard deviation values were 4.37 and 3.90%, respectively. The lifetime of electrode used to analyze EA concentration was 20 ppm for continuous injections (n = 30 and n = 40). The relative standard deviation values were 4.00 and 7.23.%, respectively. This means the electrode can be used for at least 30 measurements.
3.3 Analytical applications
Two commercial samples of skin whitening cream (0.5% (w/w) of EA), #1 and #2, were tested. Table 1 shows the comparison between the measured and label values. For 10.8 ppm of sample #1 and 4.68 ppm of sample #2, the proposed method provided good recovery in the range from 99.12 to 105.85%. These results suggest that PSPEC* coupled with FIA is a reliable detection method for EA in commercial cosmetics.| Samplea | Original detected level (ppm) | Spiked level (ppm) | Found level (ppm) | Recovery (%) |
|---|---|---|---|---|
| a The label value of #1 and #2 was 0.5% (w/w). | ||||
| #1 | 10.75 ± 0.09 | 10.00 | 20.6 ± 0.5 | 99.3%±5.7 |
| 15.00 | 26.0 ± 0.3 | 101.9%±2.2 | ||
| 20.00 | 30.5 ± 0.2 | 99.1%±1.4 | ||
| #2 | 4.53 ± 0.08 | 5.00 | 9.8 ± 0.2 | 105.8%±4.8 |
| 10.00 | 14.8 ± 0.3 | 103.4%±3.5 | ||
| 15.00 | 19.6 ± 0.2 | 100.6%±1.7 |
Conclusions
The selective determination of EA in skin whitening creams by PSPCE* coupled with FIA is presented in this work. Without the separation technique and the use of organic eluents, the detection limit for EA by the proposed method is better than the results of prior reports. The advantages of PSPCE* include easy fabrication and low cost. Providing sufficient sensitivity for EA detection, oxygen functionalities of PSPCE* gave high selectivity for EA determination. PSPCE* can be used to determine free EA not only in cosmetics but also in clinical and pharmaceutical samples.Acknowledgements
The authors would like to thank the National Science Council of Taiwan for financially supporting this research under Contract No: NSC 98-2113-M-241-002 -MY3.Reference
- B. Fuhrman, N. Volkova and M. Aviram, Nutrition, 2010, 26, 359–366 CrossRef CAS.
- C. Bell and S. E. Hawthorne, J. Pharm. Pharmacol, 2008, 60, 139–144 CrossRef CAS.
- J.-Y. Bae, J.-S. Choi, S.-W. Kang, Y.-J. Lee, J. Park and Y.-H. Kang, Exp. Dermatol., 2010, 19, e182–e190 CrossRef.
- T. Osawa, A. Ide, J. D. Su and M. Namiki, J. Agric. Food. Chem., 1987, 35, 808–812 CrossRef CAS.
- H. Shimogaki, Y. Tanaka, H. Tamai and M. Masuda, Int. J. Cosmet. Sci, 2000, 22, 291–303 CrossRef CAS.
- I. Bala, V. Bhardwaj, S. Hariharan and M. N. V. Ravi Kumar, J. Pharm. Biomed. Anal., 2006, 40, 206–210 CrossRef CAS.
- D. Venkat Ratnam, V. Bhardwaj and M. N. V. Ravi Kumar, Talanta, 2006, 70, 387–391 CrossRef CAS.
- Z. Y. He and W. S. Xia, Food Chem., 2007, 105, 1307–1311 CrossRef CAS.
- G. G. Shui and L. P. Leong, J. Chromatogr., A, 2002, 977, 89–96 CrossRef CAS.
- Y. Amakura, M. Okada, S. Tsuji and Y. Tonogai, J. Chromatogr., A, 2000, 896, 87–93 CrossRef CAS.
- J.-H. Lee, J. V. Johnson and S. T. Talcott, J. Agric. Food Chem., 2005, 53, 6003–6010 CrossRef CAS.
- P. García del Moral, M. J. Arín, J. A. Resines and M. T. Díez, J. Chromatogr., B, 2007, 855, 276–279 CrossRef CAS.
- Y. Shih and J.-M. Zen, Electroanalysis, 1999, 11, 229–233 CrossRef CAS.
- K. S. Prasad, J.-C. Chen, C. Ay and J.-M. Zen, Sens, Actuators B, 2007, 123, 715–719 CrossRef.
- C.-C. Yang, A. S. Kumar and J.-M. Zen, Anal. Biochem., 2005, 338, 278–283 CrossRef CAS.
- J.-C. Chen, A. S. Kumar, H.-H. Chung, S.-H. Chien, M.-C. Kuo and J.-M. Zen, Sens. Actuators B, 2006, 115, 473–480 CrossRef.
- J.-C. Chen, H.-H. Chung, C.-T. Hsu, D.-M. Tsai, A. S. Kumar and J.-M. Zen, Sens. Actuators B, 2005, 110, 364–369.
- K. S. Prasad, G. M. and J.-M. Zen, Electr. Commun., 2008, 10, 559–563 Search PubMed.
- A. J. Saleh Ahammad, S. Sarker, M. A. Rahman and J.-J. Lee, Electroanalysis, 2010, 22, 694–700 CrossRef.
- J.-M. Zen, H. H. Yang, M.-H. Chiu, Y.-J. Chen and Y. Shih, JAOAC Int., 2009, 92, 574–579 CAS.
- M.-H. Chiu, H.-H. Yang, C.-H. Liu and J.-M. Zen, J. Chromatogr., B, 2009, 877, 991–994 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |