DOI:
10.1039/C0AY00582G
(Paper)
Anal. Methods, 2011,
3, 646-652
Received
26th September 2010
, Accepted 6th January 2011
First published on 3rd February 2011
Abstract
A copper hexacyanoferrate nanostructure was prepared on the surface of a disposable pencil graphite electrode. The copper hexacyanoferrate nanostructures showed a pair of symmetric voltammetric peaks at 0.67 V/SCE. An electrochemical comparative characterization between copper hexacyanoferrate nano and bulk structures was presented. The transfer coefficient (α) and apparent charge rate constant (Ks) were evaluated for each of copper hexacyanoferrate nano and bulk structures. The resulting electrode exhibits an excellent electrocatalytic activity for the oxidation of ceftriaxone sodium. Cyclic voltammetry and chronoamperometry were employed to characterize the response to ceftriaxone sodium that changes linearly in the concentration range from 2 to 72 μM, with a detection limit of 0.54 μM (at a signal to noise ratio of 3). Typical features of the sensor include low cost, simple preparation, fast response, good stability, selectivity, and reproducibility. It was applied to the determination of ceftriaxone sodium in pharmaceutical samples. All results obtained were compared to the recommended procedure by British Pharmacopeia.
Introduction
Studies of one dimensional (1D) nanostructures have been attracting considerable attention in research.1,2 The synthesis of nanomaterials requires an atomistic deposition process and extreme control over the deposition. Vapor deposition techniques have been used almost exclusively to produce these materials.1,2 The fact that electrochemical deposition (ED), also being an atomic deposition process, can be used to synthesize nanostructures has generated a great deal of interest in recent years. This century-old process of ED has obvious advantages such as: rapidity, low cost, free from porosity, high purity and industrial applicability.3,4 Recently use of nanostructures in electro-catalyze application was developed.5–13 Metal and semiconductor nanoparticles as sensing elements could be immobilized on the working electrode.14 The analytical application and electrochemistry of Prussian blue analogues (PBAs) were reviewed by Karyakin.15 Among the PB analogues, copper hexacyanoferrate (CuHCF) presents special characteristics due to the capability of presenting reversible redox reactions in different supporting electrolytes.16 Besides that, CuHCF presents an increased stability in physiological pH solutions when compared with PB modified electrodes,17 leading to sensors with higher operational stability.18 On the other hand, cephalosporins are widely used in clinical therapy for the treatment of sever detection because of their antibacterial and pharmacokinetic properties.19–23 The determination of cephalosporins is important not only in the field of human health for pharmacokinetic analysis but also for quality control in food and fermentation industry to check their illegal use as in food preservation, processing, introduction of antibiotics in dairy products and as fodder additives.24Cephalosporins comprise the cephem nucleus (Scheme 1) and side-chains at positions 3 and 7, which determine their properties and bioactivity.19 The electro-activity of cephalosporins in the reduction process stems from the double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in their dihydrothiazine ring. With the exception of the electrode processes resulting from potentially active groups in substituents R1 and R3 (see Scheme 1), the reduction of these antibiotics results in the elimination of R325via a mechanism in which the first one-electron transfer is the rate-determining step.26–28
C bond in their dihydrothiazine ring. With the exception of the electrode processes resulting from potentially active groups in substituents R1 and R3 (see Scheme 1), the reduction of these antibiotics results in the elimination of R325via a mechanism in which the first one-electron transfer is the rate-determining step.26–28
Very few analyses incorporate electrochemical oxidation of cephalosporins, the first reference obtained is from the work of Fabre et al.29Oxidation of the aminothiazole group, which substituted on the side-chain in position 7 of the cephem ring in some cephalosporins, was reported to enable development of a promising amperometric detection mode for liquid chromatography or possibly other flow analytical procedures.29,30Ceftriaxone sodium is a parenteral cephalosporin that displays a broad spectrum of activity against Gram-negative and Gram-positive pathogens. In the present study of electrochemical oxidation and determination of ceftriaxone, the third generation of cephalosporin is examined using an amperometric sensor based on CuCHF nanostructures. To the best of our knowledge for the first time an electrochemical determination is reported involving a low cost nanostructure sensor for assay of the ceftriaxone sodium drug. The aim of this study is to demonstrate the electrochemical behavior of ceftriaxone sodium to develop an easy and rapid amperometry method for direct detection of ceftriaxone sodium in pharmaceutical samples without any tedious pretreatment step.
Experimental
Reagents and solutions
All chemicals were analytical grade reagents and purchased from Merck. Ceftriaxone sodium standard and real samples were kindly gifted by Daana pharmaceutical Co. All solutions were prepared with doubly distilled water. This solution was prepared daily. A KNO3 solution (0.1M) was employed as the supporting electrolyte.
Apparatus
The electrochemical setup was a conventional three-electrode cell with a platinum wire as a counter electrode and a saturated calomel electrode (SCE) as the reference electrode. All potentials are quoted with respect to the SCE. Reference and counter electrodes were obtained from Azar Electrode Co., Urmia, Iran. The working electrode (2 mm diameter) was a pencil graphite electrode (PGE) purchased from Rotring (Germany). Electrochemical experimental conditions were adjusted by a potentiostat–galvanostat (PerkinElmer, EG&G 273 A) with computerized control (M 270 software). All experiments were performed at room temperature (T = 298 K).
Preparation of CuHCF nanostructure modified PGE
The CuHCF nanostructure modified PGE was prepared in two steps. First, an unpolished PGE was immersed in a solution containing 1 mM CuSO4, 0.1 M K2SO4. In order for electrodeposition of copper at the surface of PGE, nucleation and growth pulses (i1 = −50 μA, t1 = 10 ms in nucleation pulse and i2 = −1 μA, t2 = 100 ms in growth pulse) were applied. In the second step, electrodeposited copper nanostructures were derivatized by immersing in a solution containing 1 mM K3 [Fe (CN) 6] and 0.01 M KNO3 for 5 min.
An ACE C18, 3 μm (4.6 × 100 mm) HPLC column was used. The mobile phase consisted of 2 g tetradesyl ammonium bromide, 2 g tetraheptyl ammonium bromide, 50 ml acetonitrile, 55 ml phosphate buffer (pH 7), 5 ml citrate buffer (pH 5) and 440 ml water. The flow rate of the mobile phase was 1.0 ml min−1. HPLC experimental conditions were adjusted by a 515 Waters system with a Uv-Vis detector (λ = 254 nm).
Results and discussion
For the nucleation of copper at the surface of PGE an appropriate reductive pulse current must be applied in order to set the potential of electrode on the diffusion plateau of Cu2+ ions. The duration, t1, and the current, i1, of the nucleation pulse must possess certain minimum values to force nucleation. On the other hand, care should be taken so that these parameters would not exceed certain maximum threshold levels. During the growth pulse, the crystal growth has to be conducted at a slow rate, i.e. at low overvoltage. Therefore, the pulse current applied in the growth pulse must set the potential of electrode at potentials at which electrodeposition of copper should not be observed. From Fig. 1 it has been revealed that electrodeposited copper has a structure with thickness of the monolayer less than 100 nm. The density of active sites at the surface of PGE is higher than those obtained at the surface of glassy carbon and highly oriented pyrolytic graphite electrodes due to its relatively large surface area;31 therefore, while applying a very short nucleation pulse, the initially produced copper nuclei are located above the mentioned substrate and have higher surface area than the substrates. In this case, a relatively large surface area of electrodeposited copper with a thickness in nano-scale range might induce a high electrocatalytic activity for the copper deposits.
 |
| | Fig. 1
SEM images for two different PGE surfaces following the electrodeposition of Cu+2 (0.001 M) in presence of 0.1 M K2SO4 using nucleation pulse (i1 = −50 μA, t1 = 10 ms) and growth pulse (i2 = −1 μA, t2 = 100 ms). | |
Electrochemical characterization of CuHCF nanostructures
In order to study the effect of CuHCF structure on its electron transfer reaction, besides PGE modified by CuHCF nanostructure, two PGEs were modified by cyclic voltammetry and chronoamperometry techniques. The CuHCF bulk structure was constructed at the surface of PGEs. The CV of mentioned electrodes is shown in Fig. 2. The voltammetric response of the CuHCF structures was affected by potential scan rate. As shown in Fig. 3 the peak currents increased when potential scan rate increased. At low potential scan rates the peak currents were limited by kinetic parameters, therefore there was a linear relationship between peak current and potential scan rate. The electrochemical reaction which occurred at the surface of electrode can be shown as follow:
| CuK[Fe(CN)6] + e− + K+ ⇔ CuK2[Fe(CN)6] |
![Cyclic voltammetry of PGEs modified by CuHCF in 0.01 M HClO4, at scan rate 20 mV s−1: a: Cu nanostructure electrodeposited by galvanostatic double pulse technique b: Cu bulk structure electrodeposited by CV technique ([Cu+2] = 1 mM,[K2SO4] = 0.1 M, at scan rate 10 mV s−1, number of scan = 10, E1 = −0.4, E2 = −0.7 V/SCE) c: Cu bulk structure electrodeposited by CA technique ([Cu+2] = 1 mM,[K2SO4] = 0.1 M, E1 = 0.1, E2 = −0.7 V/SCE, t = 1 s).](/image/article/2011/AY/c0ay00582g/c0ay00582g-f2.gif) |
| | Fig. 2
Cyclic voltammetry of PGEs modified by CuHCF in 0.01 M HClO4, at scan rate 20 mV s−1: a: Cu nanostructure electrodeposited by galvanostatic double pulse technique b: Cu bulk structure electrodeposited by CV technique ([Cu+2] = 1 mM,[K2SO4] = 0.1 M, at scan rate 10 mV s−1, number of scan = 10, E1 = −0.4, E2 = −0.7 V/SCE) c: Cu bulk structure electrodeposited by CA technique ([Cu+2] = 1 mM,[K2SO4] = 0.1 M, E1 = 0.1, E2 = −0.7 V/SCE, t = 1 s). | |
At potential anodic scan, K+ cations left the modifier film and entered the solution, and at potential cathodic scan, K+ cations left the solution and entered the modifier film. Hence at high potential scan rates peak currents were limited by diffusion of K+ cations. In the case of surface electrochemical reactions, ΔEp must be 0.00 V. The difference between theoretical and experimental values (reported in Table 1) can be attributed to
- Chemical interaction between ions and modifier film
- Electrostatic parameters
- Non electroactive sites in modifier film
- Kinetic limitations
Therefore, with an increase in scan rate, ΔEp increased. The Laviron equation (eqn (1)) described this relationship.32
| |  | (1) |
It is important to say in the Laviron equation ΔEp must be greater than 200/n mV.32 From Fig. 3 it was revealed that at v = 1500 mV s−1, ΔEp is greater than 200/n mV. The other symbols in (eqn (1)) have their usual meanings. Values of transfer coefficient (α) and apparent charge rate constant (Ks) which is calculated from the Laviron equation are reported in Table 1. The values of α and Ks revealed that the electron can be transferred via the CuHCF nanostructures more quickly than the bulk structures. High surface area and fast electron transfer can induce high electrocatalytic activity for CuHCF nanostructures.
Table 1 Effect of CuHCF structures on electron transfer reaction
|
Copper structure |
α
|
K
s (s−1) (n = 3) |
% RSD of Ks (s−1) |
|
Nanostructure, electrodeposited by double pulse technique |
0.52 |
2.85 |
2.46 |
| Bulk structure, electrodeposited by cyclic voltammetry technique |
0.60 |
1.50 |
3.8 |
| Bulk structure, electrodeposited by chronoamperometry technique |
0.63 |
0.93 |
3.4 |
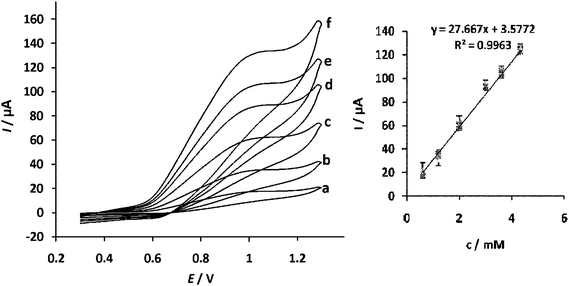
Fig. 4 shows cyclic voltammograms for the electro-catalytic oxidation of ceftriaxone sodium at the bare and modified PGEs in acetate buffer (pH 6). Upon the addition of 3 mM ceftriaxone sodium, there is a dramatic enhancement of the anodic peak current and the cathodic peak current disappeared which indicates a strong catalytic effect. The anodic peak potential for the oxidation of ceftriaxone sodium at CuHCF nanostructure PGE is about 1000 mV while at the bare electrode the ceftriaxone sodium does not have any electrocatalytic response. The dependence of surface CuHCF amount on the response of sensor was studied. From the obtained results it was revealed that the sensitivity of the sensor was influenced by the amount of electrocatalyst. When the thickness of modifier film was less than 100 nm the sensitivity of the sensor increased. Fig. 5 shows the dependence of the voltammetric response of modified PGE on the ceftriaxone sodium concentration with the addition of ceftriaxone sodium. A plot of icatvs. ceftriaxone sodium concentration was linear in the concentration range 0.6–4.5 mM. By recording cyclic voltammograms of 0.6 mM ceftriaxone sodium solution at different scan rates (data not shown), the peak currents for the anodic oxidation of ceftriaxone sodium are proportional to the square root of the scan rate. This result indicates that at sufficiently positive potential the reaction is controlled by diffusion of ceftriaxone sodium, which is the ideal case for quantitative applications. It has been reported33,34 that aminothiazole moiety can be oxidized and an anodic peak can be generated. The anodic peak appearing in the voltammogram can be related to the oxidation of this functional group.
 |
| | Fig. 5
Cyclic voltammograms of a CuHCF nanostructure modified PGE in 0.1 M acetate buffer solution (pH 6) containing 0.1 M KNO3 as supporting electrolyte, at scan rate 20 mV s −1 with various concentration of ceftriaxone sodium from 0.6–4.5 mM (a–f). | |
Electrocatalytic characteristics of ceftriaxone sodium oxidation at the modified PGE
In order to get the information about the rate-determining step a Tafel plot was drawn, using the data derived from the rising part of the current voltage curve recorded at scan rate 10 mV s −1. (Fig. 6). A slope of 61.41 mV decade −1 was obtained, indicating a one electron process was involved in the rate limiting step, assuming a charge transfer coefficient of α = 0.64. The Tafel slope (b) was also obtained from the linear relationship observed for Epvs. logv by using the following equation.35| | | Ep = (b/2) logv + constant. | (2) |
The resulting b value was obtained as 120.82 mV, which correlates with the corresponding value evaluated from polarization measurement. The catalytic oxidation of ceftriaxone sodium at the surface of the modified electrode was also studied by chronoamperometry using the potential step technique. The well defined chronoamperograms recorded on the modified electrode in the presence of ceftriaxone sodium in the range of 5 to 20 mM are illustrated in Fig. 7. For an electroactive material with diffusion coefficient D, the current corresponding to the electrochemical reaction (under diffusion control) is described by Cottrell's law:36
| | | I = nFAD1/2cπ−1/2t−1/2 | (3) |
where
D and
c are the diffusion coefficient (cm
2 s
−1) and bulk concentration (mol cm
−3) of
ceftriaxone sodium, respectively, and the other symbols have their usual meanings. The average value of
D was calculated to be 2.3 × 10
−6 cm
2 s
−1, compared with the previously reported value.
33
 |
| | Fig. 7 Chronoamperograms obtained at a CuHCF nanostructure modified PGE in the presence of 5–20 mM (a → d) ceftriaxone sodium and 0.1 M KNO3 as supporting electrolyte. The potential step was 1200 mV versusSCE. | |
 |
| | Fig. 8 Amperometric response of the CuHCF nanostructure modified PGE obtained during successive additions from ceftriaxone sodium of 0.1 mM to continuous stirring of 10 ml of 0.1 M acetate buffer solution (pH 6) containing 0.1 M KNO3 with different increments. The inset in the panel is the calibration graphs derived from the corresponding amperograms. Applied potential: 1000 mV. | |
Table 2 Comparison of major characteristics of some analytical methods used in the determination of ceftriaxone sodium
Stability of CuHCF nanostructure modified PGE
Repetitive redox cycling experiments were done to determine the extent of stability relevant to CuHCF nanostructure modified PGE in 0.1 M KNO3 solution. The results indicated that after 100 continuous cycles at 50 mV s −1, the peak heights of the CVs decreased less than 5%. On the other hand, the CuHCF nanostructure kept its initial activity for more than 2 months when exposed to air at ambient conditions. Decreases of 5 and 8% were observed in current response of the electrode at the end of 30th and 60th days, respectively.
Effect of electroactive interferences
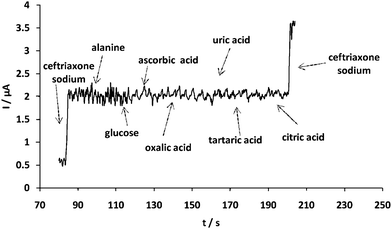
For investigating the interference, several elecrtoactive compounds were selected. If the tolerance limit was taken as the maximum concentration of the foreign substances, which caused an approximately +5% relative error, for 2 μM ceftriaxone sodium, no interference was observed for the following compounds (μM): alanine (600), glucose (600), ascorbic acid (600), oxalic acid (600), uric acid (600), tartaric acid (600), citric acid (600) (Fig. 9). The current responses generated due to these interfering species are negligible, indicating high selectivity of the sensor.
As a practical use, the sensor was also used to detect ceftriaxone sodium in pharmaceutical samples. Obtained electroanalytical results were checked by standard HPLC method (Fig. 10) for assay of ceftriaxone sodium according to British Pharmacopiea.44 These results (Table 3) suggest that the mentioned method is reliable and sensitive with regard of determining ceftriaxone sodium.
| Sample |
%Purity of ceftriaxone Sodium (n = 7) |
% RSD By proposed method |
| By proposed method |
By HPLC standard method |
| Bulk Drug |
83.75 |
84.20 |
3.5 |
| Vial 250 mg |
83.41 |
83.80 |
4.1 |
| Vial 500 mg |
83.10 |
84.05 |
3.7 |
| Vial 1g |
84.25 |
83.60 |
3.2 |
Conclusion
The new amperometric sensor developed for the determination of ceftriaxone sodium is very rapid, reproducible, highly selective and sensitive, and can be used for real sample analysis. The importance of the sensor is its noticeable characteristics which arise from the nano scale structure of the electrocatalyst which is prepared at the surface of the electrode. The independency of the system from the interferences is the feature of the technique. Furthermore, the kinetic parameters of the system calculated from the experimental results are in good agreement with those in the literature. The detection method is successfully applied for the determination of ceftriaxone sodium in the pharmaceutical samples.
Acknowledgements
We gratefully acknowledge financial support from the Post Graduate Office of the University of Tabriz and technical support of Daana pharmaceutical Co.
References
- S. C. Tjong and H. Chen, Mat. Sci. Eng., 2004, R 45, 1 Search PubMed.
- S. V. N. T. Kuchibhatla, A. S. Karakoti, D. Bera and S. Seal, Prog. Mater. Sci., 2007, 52, 699 CrossRef CAS.
- I. Gurrappa and L. Binder, Sci. Technol. Adv. Mater., 2008, 9, 1.
- L. P. Bicelli, B. Bozzini, C. Mele and L. D'Urzo, Int. J. Electrochem. Sci., 2008, 3, 356 Search PubMed.
- J. Riu, A. Maroto and F. X. Rius, Talanta, 2006, 69, 288 CrossRef CAS.
- A. Vaseashta and D. Dimova-Malinovska, Sci. Technol. Adv. Mater., 2005, 6, 312 CrossRef CAS.
- N. Atta, M. El-Kady and A. Galal, Anal. Biochem., 2010, 400, 78 CrossRef CAS.
- S. Fei, J. Chen, G. S.YaoDeng, D. He and Y. Kuang, Anal. Biochem., 2005, 339, 29 CrossRef CAS.
- D. Knopp, D. Tang and R. Niessner, Anal. Chim. Acta, 2009, 647, 14 CrossRef CAS.
- L. Agui, P. Yanez-Sedeno and J. M. Pingarron, Anal. Chim. Acta, 2008, 622, 11 CrossRef.
- C. Hierold, A. Jungen, C. Stampfer and T. Helbling, Sens. Actuators, A, 2007, 136, 51 CrossRef.
- M. Pumera, S. Sanchez, I. Ichinose and J. Tang, Sens. Actuators, B, 2007, 123, 1195 CrossRef.
- J. Qiu, W. Zhou, J. Guo, R. Wang and R. Liang, Anal. Biochem., 2009, 385, 264 CrossRef CAS.
- F. Wang and S. Hu, Microchim. Acta, 2009, 165, 1 CrossRef CAS.
- A. A. Karyakin, Electroanalysis, 2001, 13, 813 CrossRef CAS.
- D. R. Shankaran and S. S. Narayanan, Fresenius J. Anal. Chem., 1999, 364, 686 CrossRef CAS.
- R. Garjonyte and A. Malinauskas, Sens. Actuators, B, 1998, 46, 236 CrossRef.
- R. Garjonyte and A. Malinauskas, Sens. Actuators, B, 1999, 56, 93 CrossRef.
- P. Garzone, J. A. Lyon and V. L. Yu, Drug. Intell. Clin. Pharm., 1983, 17, 507 Search PubMed.
- P. Garzone, J. A. Lyon and V. L. Yu, Drug. Intell. Clin. Pharm., 1983, 17, 615 Search PubMed.
- D. S. Reeves, M. J. Bywater, D. W. Bullock and H. A. Holt, J. Antimicrob. Chemother., 1980, 6, 647 CrossRef CAS.
- T. Kamimura, Y. Matsumoto, N. Okada, Y. Mine, M. Nishida, S. Goto and S. Kuwahara, Antimicrob. Agents Chemother., 1979, 16, 540 CAS.
- K. P. Fu and H. C. Neu, Antimicrob. Agents Chemother., 1980, 17, 583 CAS.
- N. V. Shvedene and S. V. Borovskaya, J. Anal. Chem., 2003, 58, 1085 CrossRef CAS.
- M. Gchiai, O. Aki, A. Morimoto, T. Okada, K. Shinozaki and Y. Asahi, J. Chem. Soc. Perkin. Trans., 1974, 1, 258 Search PubMed.
- D. A. Hall, J. Pharm. Sci., 1973, 62, 980 CrossRef CAS.
- D. A. Hall, D. M. Berry and C. J. Schneider, J. Electroanal. Chem., 1977, 80, 155 CrossRef CAS.
- E. Munoz, J. L. Avila, L. Camacho, J. E. Cosano and F. Garcia-Blanco, J. Electroanal. Chem., 1988, 257, 281 CrossRef CAS.
- H. Fabre, M. D. Blanchin and U. Tjaden, Analyst, 1986, 111, 1281 RSC.
- H. Fabre, M. D. Blanchin and W. Kok, Analyst, 1988, 113, 651 RSC.
- M. R. Majidi, K. Asadpour- Zeynali and B. Hafezi, Electrochim. Acta, 2009, 54, 1119 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19 CrossRef CAS.
- S. Majdi, A. Jabbari, H. Heli, H. Yadegari, A. A. Moosavi-Movahedi and S. Haghgoo, J. Solid State Electrochem., 2009, 13, 407 CrossRef CAS.
- S. A. Ozkan, B. Uslu and P. Zuman, Anal. Chim. Acta, 2002, 457, 265 CrossRef CAS.
- J. A. Harrison and Z. A. Khan, J. Electroanal. Chem., 1970, 28, 131 CrossRef CAS.
-
A. J. Bard, L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley, New York, 2001 Search PubMed.
- W. Zhao, Zhang and Q. Li, Clin. Chim. Acta, 2008, 391, 80 CrossRef CAS.
- G. A. Saleh, S. R. El-Shaboury, F. A. Mohamed and A. H. Rageh, Spectrochim. Acta, Part A, 2009, 73, 946 CrossRef.
- R. Ojani, J. B. Raoof and S. Zamani, Talanta, 2010, 81, 1522 CrossRef CAS.
- D. Zhang, Y. Ma, M. Zhou, L. Li and H. Chen, Anal. Sci., 2006, 22, 183 CrossRef CAS.
- M. A. Omar, O. H. Abdelmageed and T. Z. Attia, Talanta, 2009, 77, 1394 CrossRef CAS.
- B. C. McWhinney, S. C. Wallis, T. Hillister, J. A. Roberts, J. Lipman and J. P. J. Ungerer, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2039 CrossRef CAS.
- S. Al-Rawithi, R. Hussein, D. A. Raines, I. AlShowaier and W. Kurdi, J. Pharm. Biomed. Anal., 2000, 22, 281 CrossRef CAS.
-
British Pharmacopoeia, HM Stationery Office, London, 2007.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.