Determination of Co-Q10 in plasma samples by dual-electrode amperometric detection and liquid chromatography
Megan K.
Dorris
and
Craig E.
Lunte
*
R. N. Adams Institute for Bioanalytical Chemistry, Department of Chemistry, University of Kansas, 2030 Becker Drive, Lawrence, Kansas 66047, USA
First published on 26th October 2010
Abstract
Co-Q10 is a lipid-soluble benzoquinone that is an important factor in free radical scavenging, mitochondrial membrane stability and ATP synthesis. Dietary Co-Q10 is a powerful antioxidant that has been useful in lessening the damage associated with ischemia-reperfusion injuries and aiding in the recovery of myocardial function after myocardial infarction. However, the role of dietary Co-Q10 in oxidative damage and repair is not well understood. Previous LC-EC methods have used packed carbon bed electrodes with high overpotentials that were sufficient to oxidize and reduce several biological compounds, thereby decreasing the selectivity that can be achieved with EC detection. Thin-layer cell dual electrode detection enables monitoring of reduced and oxidized forms of Co-Q10 simultaneously and selectively. The oxidation (+0.45 V vs.Ag/AgCl) and reduction (−0.4 V vs.Ag/AgCl) electrode potentials were optimized to oxidize and reduce the electroactive quinone moiety. The reduced form of Co-Q10 was prepared from the commercially available oxidized form using a Jones reductor. Confirmation of its formation was determined using the current ratios of the peak and half wave potentials from previously generated hydrodynamic voltammograms, using the oxidized form with electrodes in a series configuration. This analytical system was successfully applied to determine basal concentrations of oxidized (510 nM) and reduced (500 nM) Co-Q10 in human plasma. Peak identity of oxidized and reduced Co-Q10 was confirmed by two orthogonal methods: by the current ratios at +0.45 V and +0.25 V and −0.4 V and −0.2 V (vs.Ag/AgCl) as well as by retention time. Detection limits were determined to be 5 nM, with a linear range of three orders of magnitude.
Introduction
Ubiquinone-10 or more commonly, Coenzyme Q10 (Co-Q10), is a lipid-soluble benzoquinone that acts as an essential component for the transfer of electrons and protons between enzymatic complexes within the mitochrondrial respiratory chain.1–3 The electron transport through these complexes is required for the generation of a transmembrane proton gradient, which ultimately drives cellular energy production through ATP formation. Under oxidative stress conditions, NADH and glycerol-3-phosphate increase delivery of electrons to the enzymatic complexes within the respiratory chain resulting in an increased transfer of electrons from Complexes I and II to Complex III, which results in a high membrane potential across the mitochondria suppressing electron transfer at Complex III.4 This ultimately leads to a decrease in the production of ATP within the mitochondria and an increase in transfer of electrons to molecular oxygen elevating the production of reactive oxygen species (ROS).3,4 In addition ubiquinone-10 has also been proposed to aid in the regeneration of other endogenous antioxidants, as well as biologically regenerate itself through a series of one electron reductions making it key in the prevention of lipid peroxidation and DNA damage as a result of oxidative stress. Dietary Co-Q10 has been used as an exogenous antioxidant administered to rats who are subsequently subjected to ischemia-reperfusion. It was shown that rats administered dietary Co-Q10 showed a decrease in oxidative damage.5 In addition, dietary Co-Q10 has been shown to: reduce the depletion of ATP and the incidences of reperfusion arrhythmias, aid in the protection of membrane tissues, protect DNA and proteins against ROS, and limit the amount of lipid peroxidation and regenerate the antioxidant vitamin E (α-tocopherol).6–9Fig. 1 shows the mechanism for the reversible redox chemistry that occurs between ubiquinone-10 and ubiquinol-10.10,11 Due to the rapid redox cycling between ubiquinol-10 and ubiqinone-10 within the mitochondrial respiratory chain during oxidative stress, a method that can selectively and simultaneously detect ubiquinone-10 and ubiquinol-10 is necessary to establish the roles of this antioxidant in the maintenance of tissue homeostasis under stressful conditions.
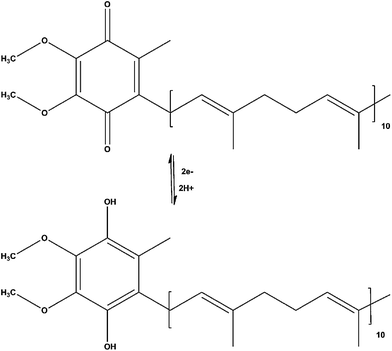 | ||
| Fig. 1 Mechanism of the electron-proton transfer of ubiquinone-10 and ubiquinol-10. | ||
Several methods have been developed for the simultaneous detection of ubiquinone-10 and ubiquinol-10 using liquid chromatography coupled with UV absorbance, fluorescence or electrochemical detection (EC). UV detection offers a simple and robust means to quantify analytes of interest, but requires significant sample pretreatment prior to analysis. In addition, ubiquinone and ubiquinol absorb at non-selective wavelengths (e.g. 254 nm) ,12–14 which in complex biological matrices, such as plasma, may result in interferences decreasing method selectivity. While UV detection is typically associated with low micromolar detection limits (∼0.5 μM); however, as Finckh et al. demonstrated, low nanomolar detection limits (∼4 nM) using UV absorbance can be obtained provided a sufficiently large sample volume is available.15 Alternatively, fluorescence detection offers a selective means to quantify ubiquinone-10 and ubiquinol-10 with low nanomolar detection limits (∼3.0 nM).16 However, sample derivatization prior to analysis is required. It has been shown that oxidation of ubiquinol-10 begins to occur within 1–2 h; therefore, any pre-treatment of the sample may result in conversion of ubiquinol-10 to ubiquinone-10, making the quantification of ubiquinol-10 inaccurate.17
EC detection provides direct and selective detection of ubiquinone-10 and ubiquinol-10. This detection technique requires no sample derivatization and is selective for analytes which are inherently electrochemically active. Previous techniques have used packed carbon bed electrodes for detection, in which a generator electrode was placed upstream from the analytical cell at a potential sufficient to completely oxidize (+0.7 V vs.Ag/AgCl) or reduce (−0.7 V vs.Ag/AgCl) the analyte of interest prior to the subsequent re-oxidation or reduction within the analytical cell10,11,15,17–28 In addition, this detection scheme contributes to increased dead volumes and residence times, resulting in increased retention times and band broadening.29 Furthermore, this detection scheme requires large sample volumes in order to overcome the large background noise associated with the increased surface area of these packed bed electrodes to obtain the low nano-molar to pico-molar detection limits typically associated with EC detection. Reduction and oxidation potentials frequently employed by these methods are known to be sufficient for the reduction of various solvents, while simultaneously oxidizing several other biologically relevant compounds often found in plasma or tissue samples decreasing the method selectivity and sensitivity. Alternatively thin layer flow cells improve mass transport efficiencies due to decreased electrode surface area and channel thickness.30 Therefore, the development of an analytical method with enhanced selectivity to qualitatively assess analytes of interest is necessary to further investigate the role of Co-Q10 in its ability to maintain tissue homeostasis during oxidative stress.
This report describes the development of an analytical method using liquid chromatography with amperometric detection for the simultaneous detection of ubiquinone-10 and ubiquinol-10 in human plasma. The method discussed offers decreased total analysis time when compared to previous methods, while maintaining low nanomolar detection limits.
Experimental
Reagents and standard preparation
Coenzyme Q10 (HPLC > 98%), zinc dust (<10 μm), lithium perchlorate (95%+) were purchased from Sigma (St. Louis, MO, USA). Sodium acetate and glacial acetic acid (17 M) used for the liquid chromatograph system were purchased from Fisher Scientific (Fair Lawn, NJ, USA). All chemicals were reagent grade and used as received. Solvents used for mobile phases or sample matrices (MeOH, EtOH, 1-propanol, hexane, acetonitrile) were all HPLC grade and were obtained from Fisher Scientific (Fair Lawn, NJ, USA). Ringer's solution consisted of 147 mM NaCl, 3 mM KCl, 1 mM MgCl2, 1.3 mM CaCl2 (152.3 mM ionic strength, Fisher Scientific, Fair Lawn, NJ, USA) and filtered through a 47 mm, 0.22 μm nylon filter (Fisher Scientific, Fair Lawn, NJ, USA) prior to use.Standard solutions were prepared by dissolving 0.4–1.5 mg of ubiquinone-10 in 1.0 ml, 1-propanol:H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). Standard solutions of ubiquinol-10 were prepared from the corresponding ubiquinone-10 using a Jones reductor. The Jones reductor consisted of zinc dust activated with dilute sulfuric acid (0.5 M). Zinc dust (1.2–1.5 g) was placed into a 3 ml BD syringe (BD Inc., Franklin Lakes, NJ, USA) with a Millex GP 0.22 μm filter with a PES membrane (Carrigtwohill, Co. Cork, Ireland). Dilute sulfuric acid (2.0 ml) was flushed through the syringe to activate the zinc, followed by rinsing with an equal volume of water. An additional 1.0 ml of acid was added and flushed through immediately before the addition of the ubiquinone-10 standard (700 μM-1.2 mM) to ensure the reaction went to completion. Any residual ubiquinol-10 was rinsed with 250 μl 1-propanol:H2O (9:1 v/v) and was collected in a 1.5 ml microcentrifuge tube. Standards were stored in the dark at −20 °C through out the day. Reductors were rinsed with 1.0 ml of water and then reactivated using the same procedure described above for multiple uses. A consistent decrease in peak height of ubiquinol-10 (∼10%) was observed when compared to the peak height of the same concentration of ubiquinone-10. It was determined that the decrease in peak height of ubiquinol-10 was due to loss of product within the reductor and not due to the formation of ubiquinone-10 as a result of oxidation. The reuse of each reductor, for multiple reactions, was determined as well as the reproducibility of the reaction from reductor to reductor was evaluated. Deviation observed from reductor to reductor was selectively eliminated based on two criteria. First, solution color was taken into account since this has been reported previously as a means to determine formation of the quinol.17 Secondly, chromatographic verification was used to ensure that there was no quinone present. Only those reductors which produced a clear solution, as well as no chromatographic evidence of the quinone were used for future method development. The relative standard deviations, based on peak heights between reductors, as well as multiple uses of a single reductor, were determined to be 6.6% and 6.2%, respectively. It was concluded that no significant variation occurred in the preparation of each reductor to notably affect the reduction of ubquinone-10 to ubiquinol-10.
:1 v/v). Standard solutions of ubiquinol-10 were prepared from the corresponding ubiquinone-10 using a Jones reductor. The Jones reductor consisted of zinc dust activated with dilute sulfuric acid (0.5 M). Zinc dust (1.2–1.5 g) was placed into a 3 ml BD syringe (BD Inc., Franklin Lakes, NJ, USA) with a Millex GP 0.22 μm filter with a PES membrane (Carrigtwohill, Co. Cork, Ireland). Dilute sulfuric acid (2.0 ml) was flushed through the syringe to activate the zinc, followed by rinsing with an equal volume of water. An additional 1.0 ml of acid was added and flushed through immediately before the addition of the ubiquinone-10 standard (700 μM-1.2 mM) to ensure the reaction went to completion. Any residual ubiquinol-10 was rinsed with 250 μl 1-propanol:H2O (9:1 v/v) and was collected in a 1.5 ml microcentrifuge tube. Standards were stored in the dark at −20 °C through out the day. Reductors were rinsed with 1.0 ml of water and then reactivated using the same procedure described above for multiple uses. A consistent decrease in peak height of ubiquinol-10 (∼10%) was observed when compared to the peak height of the same concentration of ubiquinone-10. It was determined that the decrease in peak height of ubiquinol-10 was due to loss of product within the reductor and not due to the formation of ubiquinone-10 as a result of oxidation. The reuse of each reductor, for multiple reactions, was determined as well as the reproducibility of the reaction from reductor to reductor was evaluated. Deviation observed from reductor to reductor was selectively eliminated based on two criteria. First, solution color was taken into account since this has been reported previously as a means to determine formation of the quinol.17 Secondly, chromatographic verification was used to ensure that there was no quinone present. Only those reductors which produced a clear solution, as well as no chromatographic evidence of the quinone were used for future method development. The relative standard deviations, based on peak heights between reductors, as well as multiple uses of a single reductor, were determined to be 6.6% and 6.2%, respectively. It was concluded that no significant variation occurred in the preparation of each reductor to notably affect the reduction of ubquinone-10 to ubiquinol-10.
Chromatographic system
All samples were analyzed by LC-EC. The system consisted of a Shimadzu LC-20AD pump and electrochemical detection was achieved with a Bioanalytical Systems LC-4C dual-electrode amperometric detector using glassy carbon electrodes (Bioanalytical Systems Inc., West Lafayette, IN). A Ag/AgCl reference electrode (3 M NaCl) was used and all potentials are reported versus this electrode. Sample injections were made using a Rheodyne model 7125i injector (overfill of 25 μl into a 5 μl PEEK sample loop). Data was acquired using Chrom and Spec software, Chromatography Data System, version 1.5x (Ampersand International, Inc., Cleveland, OH, USA).Separation was achieved using a Zorbax Bonus RP C18 column (2.1 × 100 mm, 3.5 μm) (Agilent Technologies, Santa Clara, CA, USA). The mobile phase consisted of MeOH/ACN/glacial acetic acid (59/39/2% v/v) with 55 mM sodium acetate at a flow rate of 0.75 ml/min. Oxygen was removed from the system by continuously purging the mobile phase with argon.
Sample preparation
Pooled plasma (Invitrogen, Carlsbad, CA, USA) (100 μl) was transferred into a 1.5 ml microcentrifuge tube. Extraction of ubiquinone-10 and ubiquinol-10 was performed as previously reported by Kalenikova.7 Briefly, 100 μl of plasma was mixed with 200 μl ethanol and 550 μl hexane and shaken for 10 min, followed by centrifugation at 6,700 rpm for 3 min. The analytes were extracted from the EtOH using hexane. The hexane layer was removed (500 μl) and an additional 550 μl of hexane was used to wash the remaining sample. The process was repeated, and the excess hexane (500 μl) layer was added to the first aliquot with a final volume of 1.5 ml. The hexane was then evaporated to dryness under argon and reconstituted with 100 μl 1-propanol:H2O (9:1 v/v), and immediately analyzed. Standard additions were used to determine the basal concentration of ubiquinone-10 and ubiquinol-10 in plasma samples. The concentrations determined using standard additions were then compared to basal concentrations, from unspiked plasma samples, whose concentrations were determined using standard curves. The ratio of the basal concentrations determined from the standard additions were then compared to those determined from the linear regression analysis to determine the extraction efficiency for ubiquionone-10 and ubiquinol-10 from the pooled human plasma samples.
Results and discussion
Method development
Ubiquinone-10 has both polar and non-polar functionalities, resulting in interactions with either normal or reverse phase systems. Conventionally, normal phase systems would be a logical choice for the separation of bulky lipophilic compounds, such as ubiquinone-10, due to the minimal interactions that would be expected with the stationary phase. Columns used for normal phase systems are typically silica based columns. Under normal phase conditions it has been observed that compounds such as ubiquinone, strongly adsorb on the stationary phase lowering efficiencies when compared to reverse phase systems. For these reasons, reverse phase separations with non-aqueous mobile phases are frequently employed.Previously developed methods have used mobile phases consisting of a wide variety of solvents (e.g.MeOH, EtOH, 1-propanol, IPA, etc.); however, these solvents often contain several trace elements that generally result in increased background noise and correspondingly poorer limits of detection. Initially, for the separation of ubiquinone-10 and ubiquinol-10 a mobile phase consisting of ACN:MeOH (75:25 v/v) with 50 mM lithium perchlorate, (utilizing a Phenomenex Gemini RP-C18, 150 × 2.0 mm, 5 μm) was employed as described by Tang et al.10 Under these conditions a total analysis time of 25 min was obtained with peak widths of over a minute. In order to improve efficiencies and decrease band broadening different ratios of ACN and MeOH were investigated. It was determined that varying the amount ACN and MeOH in the mobile phase had no significant effect on retention time or peak efficiencies. Therefore, the stationary phase was investigated to determine if changing the hydrophobicity of the column packing would decrease retention times and improve band broadening. The RP-C18 Zorbax Bonus-RP (3.5 μm, 2.1 × 100 mm) provided a triply end-capped stationary phase, minimizing non-polar interactions with the incorporation of a polar amide group in the alkyl chain, modifying the interaction of ubiquinone-10 with the stationary phase, when compared to traditional RP-C18 columns, which expose long chain hydrocarbons resulting in increased interactions with the poly-unsaturated backbone of ubiquinone-10. Using this column reduced total analysis times from 25 min to 8 min, while also decreasing peak width from ∼1.5 min to 0.62 min, respectively. The ratios of ACN and MeOH were subsequently varied to further decrease peak width and improve retention times. It was determined that a mobile phase consisting of 60:40 (% v/v) MeOH:ACN proved to give the optimal separation conditions by further reducing the retention times of ubiquinone-10 and ubiquinol-10 to 5 and 4 min, respectively. Table 1 summarizes the key parameters determined for this method. Although, there is a decrease in peak efficiencies from the initial conditions using a Gemini RP C-18 (N = ∼6600) to the Zorbax Bonus RP (N = ∼1600), column interactions were minimized resulting in decreased retention times and band broadening was reduced.
Although lithium perchlorate is a typical inorganic salt used in non-aqueous mobile phases, it was observed that over time precipitation of the salt from the mobile phase was accumulating at the surface of the electrodes resulting in cross-talk. Substitution of lithium perchlorate by sodium acetate (55 mM) eliminated the precipitation and resulting cross-talk while providing sufficient conductance to minimize noise.
As discussed previously, ubiquinol-10 is readily oxidized to ubiquinone-10 when exposed to light at room temperature. In addition, oxidation to the respective quinone moiety also occurs in the presence of oxygen. In order to minimize loss of target analyte, glacial acetic acid (2% v/v) was added to the mobile phase described above reducing the pH to ∼6. The addition of acetic acid in the mobile phase provided supplementary protons in solution reducing any potential oxidation of ubiquinol-10 to ubiquinone-10.
Electrochemical detection
Detection potentials for both electrodes were determined from hydrodynamic voltammograms (HDV), as shown in Fig. 2. HDV's were determined by making multiple injections of a 2 μM standard of ubiquinone-10 or ubiquinol-10 and stepping the potential in 50 mV increments between injections. Peak heights were normalized, to the largest response in order to compare chromatograms for direct and indirect detection of ubiquinone-10 and ubiquinol-10.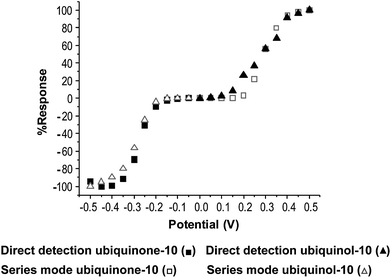 | ||
| Fig. 2 Comparison of HDV's obtained directly and in the series configuration for ubiquionone-10 (■,□) and ubiquinol-10 (▲,△). | ||
Series dual electrode configuration
In the series configuration HDV's can be generated at the downstream electrode of the product generated at the upstream electrode. Initially, unbiquinol-10 was injected while the upstream electrode was held at +0.45 V and the downstream electrode had an applied potential of 0.0 V. For each subsequent injection of ubiquinol-10 the downstream potential was stepped down incrementally from 0.0 V to −0.50 V. Next, ubiquinone-10 was injected and the upstream electrode was held at −0.40 V, while the downstream electrode was again held at 0.0 V. Upon subsequent injections of ubiquinone-10 the downstream electrode was stepped 50 mV in the positive direction, until +0.50 V was reached. Based on the HDV, shown in Fig. 2, detection potentials were established to be +0.45 V and −0.40 V. These potentials were chosen, because the largest analytical signal is obtained at these potentials while generating the least amount of noise.In the series configuration selectivity is enhanced due to the ability to monitor chemically reversible processes at the downstream electrode. This is advantageous as chemically reversible redox couples, such as ubiquinone-10 and ubiquinol-10, can be easily distinguished at the downstream electrode from the chemically irreversible species, which more easily oxidize or reduce at the upstream electrode. This configuration was used to confirm the formation of ubiquinol-10 from ubiquinone-10 using the Jones reductor by comparing the current ratios at the peak and half wave potentials for a standard of ubiquinol-10versus endogenous ubiquinol-10. The same experiment was performed, as mentioned above, using endogenous samples and standards to demonstrate the selectivity of this electrode configuration. Ubiquonone-10 was used as a control for this experiment. The current ratios determined for ubiquionol-10 (i+0.3/+0.45) and ubiquinone-10 (i−0.25/−0.4) standards and endogenous ubiquinol-10 and ubiquinone-10 were 0.77, 0.42 and 0.72, 0.47, respectively. This shows that the Jones reductor produced only ubiquinol-10 and further demonstrates the selectivity of this electrode configuration. A representative chromatogram for the separation of ubiquinone-10 and ubiquiol-10, with the electrodes in series configuration, is shown in Fig. 3. Good baseline resolution was obtained under these conditions, and both compounds were detected in less than 5 min, decreasing retention times previously reported10,15,21,23,24
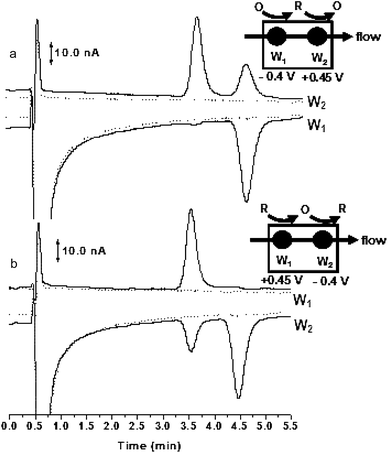 | ||
| Fig. 3 Chromatograms showing the response of ubiquinone-10 and ubiquinol-10 using the series dual-electrode configuration. 3a, reduction electrode (−0.40 V vs.Ag/AgCl) is upstream and oxidation electrode (+0.45 V vs.Ag/AgCl) is downstream; 3b, oxidation electrode (+0.45 V vs.Ag/AgCl) is upstream and reduction electrode (−0.40 V vs.Ag/AgCl) is downstream. The separation was carried out using Zorbax Bonus RP C18 column (2.1 × 100 mm, 3.5 μm) with MeOH/ACN/gacial acetic acid (59/39/2% v/v), 55 mM sodium acetate at a flow rate of 0.75 ml/min. | ||
Parallel dual electrode configuration
Although the series electrode configuration improves selectivity, when analyzing complex biological samples, the sensitivity of the method can be improved using the electrodes in the parallel configuration. For ubiquinol-10 injections started at 0.0 V and were incrementally increased to +0.5 V. Similarly, for ubiquinone injections began at 0.0 V and the potential was incrementally decreased to −0.5 V. Detection potentials for the oxidation and reduction electrodes were determined to be +0.45 V and −0.4 V, respectively.In this configuration two different potentials can be monitored for simultaneous detection of reducible and oxidizable species directly. Fig. 4 shows the response for ubiquinone-10 and ubiquinol-10versus the response of the sample matrix with the electrodes in a parallel configuration. In this configuration the analyte of interest is seen at both the oxidation and reduction electrodes simultaneously; therefore, no subsequent re-oxidation or reduction can occur, as shown in Fig. 4a and 4b. In addition, in this configuration qualitative information can be easily obtained. For the purpose of this method, this configuration qualitatively and simultaneously shows the redox state of endogenous Co-Q10, as shown in Fig. 4c, with increased sensitivity compared to previously published methods. Based on this reasoning the electrodes were used in the parallel configuration for further method validation.
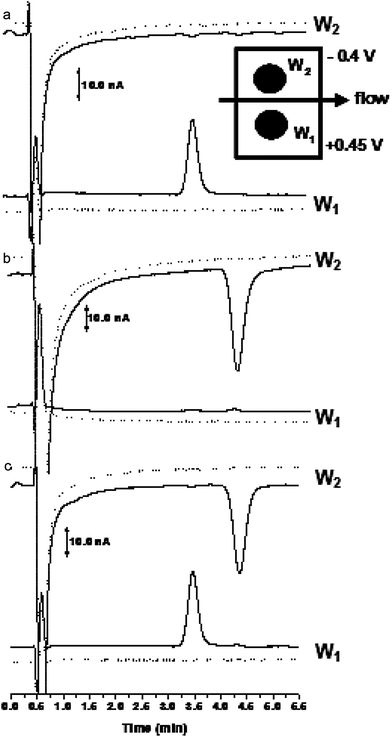 | ||
| Fig. 4 Chromatograms showing the response of ubiquinone-10 and ubiquinol-10 using the parallel configuration dual-electrode detection. Figure 4a injection of ubiquinone-10 and 4b injection of ubiquinol-10. Figure 4c injection of ubiquinone-10 and ubiquinol-10 using the parallel electrode configuration. Chromatographic conditions as listed in Fig. 3. | ||
Method validation
The calibration curves were linear over three orders of magnitude (0.05–2.0 μM), with correlation coefficients of 0.997–1. The limit of detection (LOD, S/N = 3) for this method were determined to be 5.0 nM with a RSD of 8% and 11% for ubiquinone-10 and ubiquinol-10, respectively. The limit of quantification (LOQ, S/N = 10) was subsequently determined to be 20 nM with a RSD of 5.4% and 2.2% for uniquonone-10 and ubiquinol-10, respectively. In addition, the intra-day and inter-day reproducibility of the method was determined by comparing peak areas and deviation in sensitivity. This was achieved by analyzing several different standard concentrations initially, followed by successive injections (n = 10) of two standard concentrations, which were not included in the initial calibration curve. The analysis of the peak areas was then performed and it was determined that the sensitivity of the method was consistent over the course of a day, with RSD's of 7.8% and 6.0% for 0.05 μM and 1.6% and 2.2% for 1.0 μM for both ubiquinone-10 and ubiquinol-10, respectively. Intra-day reproducibility was determined using the same method of analysis. The deviation in method sensitivity was determined to be ±6% and ±12%, for ubiquinone-10 and ubiquinol-10, respectively. The larger deviation for ubiquinol-10 was attributed the reproducibility of the Jones reductor.The stability of ubiquinol-10 in samples was determined over the course of a 24 h period. Injections of the same standard concentration (1.0 μM) were performed every hour for 8 h. Peak heights were compared to determine any relative changes in ubiquinol-10. From this analysis, it was determined that there was no significant change in peak height over the course of 8 h. The standard was then stored at −20 °C overnight in the dark and the same standard was subsequently injected the following day. From this it was determined that after a 24 h period there was significant loss in the response of ubiquinol-10 and the formation of ubiquinone-10 was observed. The loss in response of ubiquinol-10 was determined to be a result of time and not due to any freeze/thaw cycle that occurred. Therefore, standards were prepared fresh daily.
Plasma sample analysis
Fig. 5 shows a representative chromatogram from a plasma extraction. The method shows good selectivity for ubiquinone-10 and ubiquinol-10. In order to quantify ubiqunone-10 and ubiquinol-10 in plasma samples a calibration curve was generated. Standards of ubiquinone-10 and ubiquinol-10 were prepared as previously discussed. Standard concentrations ranged from 0.1– 2.0 μM, with n = 3 injections performed for each concentration. Good linearity was obtained over the desired concentration range (R2 = 0.99). The concentrations of ubiquinone-10 and ubiquinol-10, determined using the generated standard curves, were calculated to be 0.42 μM and 0.45 μM, with RSD's of 23% and 21%, respectively. Plasma concentrations reported previously range from 480 nM–2.0 μM for ubiquinol-10 and 16–900 nM for ubiquinone-10.26 These values agree well with the concentrations reported for this analysis. Previously, there has been concern about the stability of ubiquinol-10 during the extraction from plasma.10 Tang et al. concluded in a study that both the solvent used for the extraction as well as pre-column reduction were key in the recovery of ubiquinol-10 from various types of plasma; however there was no consistent trend within the data. In addition, several internal standards were tested with the optimized method, but were not included due to either elution within the void or poor capacity factors. Therefore, to validate the stability of ubiquinol-10 during extraction from plasma standard additions were performed. This was done by spiking three different known concentrations of standards into separate aliquots of plasma. Table 2 summarizes the extraction efficiencies determined for each concentration. The deviation in values was attributed to reproducibility of the extraction. Basal concentrations were found by extrapolating the best fit line across the x-axis and were determined to be 0.51 μM (±14%) and 0.50 μM (±10%) for ubiquinone-10 and ubiquinol-10, respectively. The standard additions were then compared ratiometrically to the concentrations determined from the standard curves. Based on this method of analysis, extraction efficiencies were determined to be 82% and 90% for ubiquinone-10 and ubiquinol-10, respectively. These values compare well with previously reported extraction efficiencies, specifically those reported for ubiquinol-10.10 It was therefore concluded that no significant oxidation of ubiquinol-10 occurred during extraction.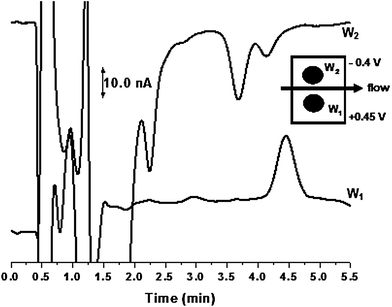 | ||
| Fig. 5 Detection of ubiquinone-10 and ubiquinol-10 in pooled human plasma using a parallel electrode configuration. Conditions as in Fig. 3. | ||
| Spiked concentration (nM) | Extraction efficiency ubiquinone-10 | Extraction efficiency ubiquinol-10 |
|---|---|---|
| a n = 6 extractions/concentration. | ||
| 500 | 99.9% ± 4.3% | 100% ± 11.6% |
| 750 | 93.9% ± 7.1% | 89.0% ± 8.0% |
| 1000 | 87.8% ± 11.7% | 85.9% ± 9.7% |
Conclusion
In conclusion, a robust and rapid analytical method with enhanced selectivity for the determination of ubiquinone-10 and ubiquinol-10 has been developed. This was achieved by using a dual glassy carbon electrode in a parallel electrode configuration. Total analysis times were improved based upon previously published methods, while maintaining good baseline resolution, peak efficiencies and method sensitivity for ubiquinone-10 and ubiquinol-10. These improvements allow for the determination of the redox status of ubiquinone-10 and ubiquinol-10, which will improve understanding of their role in maintaining tissue homeostasis as a result of an oxidative stress event.Acknowledgements
This work was supported by a grant from the National Institutes of Health (R01-NS066466).References
- M. Dhanasekaran and J. Ren, Curr. Neurovasc. Res., 2005, 2(5), 447–459 Search PubMed.
- J. I. Mechanick, Nutr. Strategies Diabetic Prediabetic Patient, 2006, 221–263 Search PubMed 1 plate.
- A. Naini, et al. , BioFactors, 2003, 18(1–4), 145–152 Search PubMed.
- G. T. Chew and G. F. Watts, QJM: Mon. J. Assoc. Physicians, 2004, 97(8), 537–48 Search PubMed.
- N. A. Khan, et al. , Int. J. Biol. Chem., 2010, 4(1), 1–9 Search PubMed.
- Y. Birnbaum, S. L. Hale and R. A. Kloner, Cardiovasc. Res., 1996, 32(5), 861–868 CAS.
- E. I. Kalenikova, et al. , Biochemistry (Moscow), 2007, 72(3), 332–338 CrossRef CAS.
- V. L. Lakomkin, et al. , Biochemistry (Moscow), 2005, 70(1), 79–84 CAS.
- V. L. Lakomkin, et al. , Biochemistry (Moscow), 2004, 69(5), 520–526 CrossRef CAS.
- P. H. Tang, et al. , Clin. Chem. (Washington, DC, U.S.), 2001, 47(2), 256–265 CAS.
- P. H. Tang and T. de Grauw, Clin. Chem. (Washington, DC, U.S.), 2004, 50(10), 1930–1932 CrossRef CAS.
- C. A. Gay and R. Stocker, Methods Enzymol., 2004, 378, 162–169 CAS (Quinones and Quinone Enzymes, Part A).
- K. Li, et al. , Biomed. Chromatogr., 2006, 20(10), 1082–1086 CrossRef CAS.
- G. Rousseau and V. F., J. Chromatogr. Sci., 1998, 36, 247–252 CAS.
- B. Finckh, et al. , Anal. Biochem., 1995, 232(2), 210–16 CrossRef CAS.
- S. Andersson, J. Chromatogr., A, 1992, 606(2), 272–6 CrossRef CAS.
- Y. Yamamoto and S. Yamashita, Methods Mol. Biol. (Totowa, NJ, U.S.), 2002, 186, 241–246 Search PubMed (Oxidative Stress Biomarkers and Antioxidant Protocols).
- M. Battino, L. Leone and S. Bompadre, Methods Enzymol., 2004, 378, 156–162 CAS (Quinones and Quinone Enzymes, Part A).
- P. O. Edlund, J. Chromatogr., Biomed. Appl., 1988, 425(1), 87–97 CrossRef CAS.
- B. Finckh, et al. , Methods Enzymol., 1999, 299, 341–348 CAS (Oxidants and Antioxidants, Part A).
- A. Galinier, et al. , FEBS Lett., 2004, 578(1–2), 53–57 CrossRef CAS.
- J. K. Lang, K. Gohil and L. Packer, Anal. Biochem., 1986, 157(1), 106–16 CAS.
- C. Leray, et al. , J. Lipid Res., 1998, 39(10), 2099–2105 CAS.
- T. Menke, et al. , Anal. Biochem., 2000, 282(2), 209–217 CrossRef CAS.
- M. Podda, et al. , Methods Enzymol., 1999, 299, 330–41 CAS.
- E. Steele Paul, et al. , Am. J. Clin. Pathol., 2004, 121(Suppl), S113–20.
- H. Tang Peter, et al. , Clin. Chim. Acta, 2004, 341(1–2), 173–84 CrossRef CAS.
- S. Yamashita and Y. Yamamoto, Anal. Biochem., 1997, 250(1), 66–73 CrossRef CAS.
- Kissinger, P. T., Heineman, W. R., ed. Laboratory Techniques in Electroanalytical Chemistry, Marcel Dekker, Inc.: New York, 2nd edn, 1996, Search PubMed.
- D. A. Roston, R. E. Shoup and P. T. Kissinger, Anal. Chem., 1982, 54(13), 1417A–1434A CAS.
| This journal is © The Royal Society of Chemistry 2011 |