SPR-based single nucleotide mismatch biosensor
Eftim
Milkani
ab,
Aung M.
Khaing
bc,
Sergi
Morais
d,
Christopher R.
Lambert
*b and
W. Grant
McGimpsey
abc
aChemistry and Biochemistry, Worcester Polytechnic Institute, 100 Institute Road, Worcester, MA 01609, USA
bBioengineering Institute, Worcester Polytechnic Institute, 100 Institute Road, Worcester, MA 01609, USA. E-mail: clambert@wpi.edu
cBiomedical Engineering, Worcester Polytechnic Institute, 100 Institute Road, Worcester, MA 01609, USA
dInstituto de Química Molecular Aplicada, Universidad Politécnica de Valencia, Camino de Vera s/n, 46071, Valencia, Spain
First published on 23rd November 2010
Abstract
The detection and characterization of the hybridization event of 21-base, unlabeled DNA oligonucleotides with a monolayer of complementary DNA immobilized on a gold surface, by electrochemical impedance spectroscopy and surface plasmon resonance (SPR) is presented. A thiol modification on the probe DNA strand allowed for its attachment to the surface viaself-assembly. For the hybridization of full match target DNA a detection limit of 20 pM was determined. RNA hybridization was also detectable with the same sensor, with a similar detection limit. The SPR signal generated upon hybridization of the full match was always distinguishable from the single mismatch target DNA oligonucleotides when the mismatch was in the middle or at the proximal end of the target DNA sequence. However, the response of the sensor was identical for the hybridization of the full match and the distal end mismatch. The SPR sensor described is reusable over at least 20 hybridization/regeneration cycles and is insensitive to flow rate (20–800 µL min−1) or temperature (20–60 °C). Based on the SPR response, the surface density of the probe was estimated to be at least 4.3 × 1012 molecules per cm2.
Introduction
Hybridization of oligonucleotides with immobilized DNA has become increasingly important as a probe for use in biosensing applications. These applications include detection of single-nucleotide polymorphisms (SNPs) and genetically modified organisms (GMOs). A SNP is a single nucleotide mutation in the genome of an organism. There are approximately 10 million variance sites or SNPs in the world's human population and they make up most of the 0.1% genetic variance observed in the human population.1 Common diseases, including cancer, are known to be caused by combinations of multiple genetic and environmental factors.2 Large-scale surveys have already associated certain common diseases with specific genes and variations in their sequence.3 There is an international effort underway to create a map of all human SNPs and determine their connections to many diseases,4 and at the same time their link to differences in response to therapeutic agents.5 GMOs are organisms whose genome has been altered by using genetic engineering, usually through the introduction of a gene from another organism. The extensive recent use of GMOs in agriculture and their increased use in the manufacture of food products require the detection of GMOs in these products for labeling and regulation purposes.6Detection of RNA is important for studying and monitoring gene expression. Of particular relevance to the present work is the detection of micro-RNAs (miRNAs). These RNAs are small, non-coding, 18 to 24-nucleotide single-stranded sequences that are involved in gene regulation, affecting essential processes such as cell proliferation, cell death, tumor genesis, and mammalian cell development.7–9 Due to their small size, miRNAs are difficult to detect using conventional methods10 and there is a pressing need for rapid, label-free detection methods. This would speed ongoing research in understanding their role in gene regulation, and potentially lead to the development of new medical therapeutic technologies which will be based on the ability of miRNAs to block and/or control gene expression.
The detection of nucleic acids requires a platform which is able to sense the hybridization process and also facilitates the immobilization of single-stranded (ss) probe DNA, while maintaining its ability to efficiently hybridize with the target sequence. Surface plasmon resonance (SPR) is a surface characterization technique that can be employed to report changes on a metal-coated surface based on alterations occurring in the dielectric medium within a very short distance (100–400 nm) from the metal surface.11SPR provides non-invasive, real time, label-free detection of biomolecular binding interactions including DNA–DNA and DNA–RNA interactions.12–16 In addition, SPR allows measurements of kinetic rates and equilibrium affinity constants for these interactions.17
SPR sensor surfaces consist of a conductive substrate or chip on whose surface a probe DNA is immobilized. Previous work on SPR detection of DNA hybridization includes surfaces that contain 100 nm thick carboxymethylated dextran polymers to which the probe DNA is anchored with biotin–streptavidin.12,18–20 The sensitivity of this kind of probe surfaces is dependent on the diffusion of the analyte. Mark et al. monitored DNA hybridization reactions with dendrimer-functionalized self-assembled monolayers (SAMs) with a detection limit of 3.9 nM,21 while Vaisocherova et al. used streptavidin-functionalized SAMs to reach a similar detection limit.22 Other researchers have monitored DNA hybridization by covalently attaching probe DNA to an amino terminated SAM.16 Lower detection limits have been reported, but they involve the use of DNA-capped gold nanoparticles.23 Another label-free optical technique is total internal reflection ellipsometry, which has been shown to detect both genomic DNA and DNA oligonucleotides.24,25 Piezoelectric methods are able to convert minute mass changes on the surface into nanomechanical response signals, and therefore they also do not require labeling of target DNA. The lowest detection limit was reported to be 10 nM.26
Detection of SNPs or single nucleotide mismatches by SPR has been reported by Nakatani et al. who showed detection of guanine–guanine mismatches by using a mismatch specific ligand.27 They were able to detect these mismatches at 1 nM oligonucleotide concentrations. The same group was also able to detect guanine–adenine mismatches down to 100 nM concentrations using a synthetic aromatic molecule which stabilized the mismatch.28 Others were able to detect mismatches using a thiolated probe DNA with a detection limit of 500 nM.29 Tawa and Knoll used fluorescent labeled target DNA sequences to distinguish SNPs using surface plasmon fluorescence spectroscopy.30 Dell'Atti et al. reported a piezoelectric biosensor with a 100 nM detection limit with regard to detection of SNPs.31
In a recent publication of preliminary work describing the SPR monitoring of oligonucleotide hybridization we reported the capability to detect single nucleotide mismatches upon hybridization of target DNA oligonucleotides to thiol-modified probe DNA oligonucleotides using an integrated, three channel SPR chip.32 In the present paper we report the further characterization of the DNA surface deposition and hybridization using electrochemistry and SPR and the analysis of SPR data to determine the surface coverage of probe DNA. The SPR sensor surface was modified with a mixed monolayer of hexanethiol-modified 21-base long probe DNA and 6-mercapto-1-hexanol (MHO) (Fig. 1). MHO was used to avoid non-specific adsorption and to facilitate hybridization of target DNA by spacing the probe oligonucleotides, as described previously.33–36 The probe DNA strand was modified with a hexanethiol tether [HS–(CH2)6−] at the 5′ end. The 21-base target oligonucleotides were not modified. Hybridization was characterized and detected by electrochemistry and SPR. It was possible to use the sensor to detect single mismatches and the signal response was found to be sensitive to the position of the mismatch. The sensor surface could be regenerated by washing the surface in situ with a denaturing solution, allowing for the sensor surface to be reused for subsequent hybridization measurements.
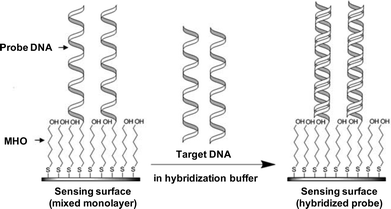 | ||
| Fig. 1 Depiction of the SPR sensing surface. The sensing surface consists of a mixed monolayer of hexanethiol-modified probe DNA oligonucleotide (probe DNA) and 6-mercapto-1-hexanol (MHO) chemisorbed on the gold surface of the SPR sensor through the sulfur of the thiol group. Upon target DNA exposure the hybridization of probe with target strands occurs resulting in a detectable change in the SPR signal. | ||
Experimental
Materials
All chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated. Potassium chloride (99.995% purity) and 35% w/w hydrogen peroxide were purchased from Alfa Aesar (Ward Hill, MA, USA), sulfuric acid from EMD Chemicals, Inc. (Gibbstown, NJ, USA), and anhydrous ethanol from Pharmco-AAPER (Brookfield, CT, USA). Phosphate buffer (PB) was prepared at pH 7.0 as a mixture of 10 mM sodium dihydrogen phosphate and 10 mM sodium hydrogen phosphate in deionized water. Hybridization buffer was prepared as a 20× concentrated stock solution (3.0 M NaCl, 0.3 M sodium citrate, pH 7.0). All aqueous solutions were prepared in deionized water obtained from a Millipore Synergy UV system (Billerica, MA, USA).All aqueous solutions used for SPR experiments were filtered prior to use through a 1 µm PURADISC™ 25 GD filter from Whatman (Florham Park, NJ, USA).
The thiol modified probe DNA oligonucleotide was obtained from Fidelity Systems, Inc. (Gaithersburg, MD, USA). Unmodified target DNA and RNA oligonucleotides were purchased from Sigma Genosys (The Woodlands, TX, USA). They consisted of one full complementary strand, one non-complementary strand, and three one-mismatch complementary strands containing a single-nucleotide mismatch in the 5′ end (P1), 3′ end (P21), or middle of the sequence (P11) (Table 1). The non-complementary strand served as a non-thiolated control probe. The RNA strands consisted of a non-complementary and a full complementary strand. The oligonucleotides were reconstituted in autoclaved deionized water, at 1 mM concentration for the thiol-modified strand and 100 µM for the unmodified target strands. RNA oligonucleotides were stored at −80 °C, whereas DNA oligonucleotides were stored at −20 °C.
| Oligonucleotide | Sequence (5′–3′) |
|---|---|
| Probe DNA (DNA–SH) | HS–(CH2)6–CCCGATTGACCAGCTAGCATT |
| Full match (FM) | AATGCTAGCTGGTCAATCGGG |
| Middle mismatch (P11) | AATGCTAGCT![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) GTCAATCGGG GTCAATCGGG |
| 5′ End mismatch (P1) |
![[G with combining low line]](https://www.rsc.org/images/entities/b_char_0047_0332.gif) ATGCTAGCTGGTCAATCGGG ATGCTAGCTGGTCAATCGGG |
| 3′ End mismatch (P21) | AATGCTAGCTGGTCAATCGG![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) |
| Control (full mismatch) | CCCGATTGACCAGCTAGCATT |
| RNA full match | AAUGCUAGCUGGUCAAUCGGG |
| RNA control (full mismatch) | CCCGAUUGACCAGCUAGCAUU |
Gold slides consisting of 1 mm float glass with a 5 nm film of chromium and 100 nm of evaporated gold were obtained from Evaporated Metal Films (Ithaca, NY, USA) and used for electrochemical measurements. Slides were cleaned in piranha solution (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v H2SO4/35% H2O2) for 10–20 min (Caution: piranha reacts violently with organic compounds and should not be stored in closed containers), rinsed several times with deionized water and finally rinsed with anhydrous ethanol. The slides were dried under a stream of nitrogen gas before cleaning in oxygen plasma for 45 seconds using a Plasma Prep II plasma cleaner (SPI Supplies, West Chester, PA, USA).
:3, v/v H2SO4/35% H2O2) for 10–20 min (Caution: piranha reacts violently with organic compounds and should not be stored in closed containers), rinsed several times with deionized water and finally rinsed with anhydrous ethanol. The slides were dried under a stream of nitrogen gas before cleaning in oxygen plasma for 45 seconds using a Plasma Prep II plasma cleaner (SPI Supplies, West Chester, PA, USA).
SPREETA SPR sensing modules, model TSPR1K23, were purchased from ICx Nomadics (Stillwater, OK, USA). The modules contain a 0.12 mm thick borosilicate glass slide (16.9 mm × 5.6 mm) coated with 500 Å of gold as the sensing surface. The gold film on the SPR sensing surface was rinsed with deionized water and anhydrous ethanol. The sensor surface was dried with a stream of nitrogen and the sensor was cleaned in oxygen plasma for 45 seconds prior to surface modification. Oxygen plasma exposure was found not to affect the performance of the sensor.
Surface modification of gold slides with probe DNA
Immediately after plasma cleaning, the gold slides were covered with a 10 µM solution of thiol-modified probe DNA (DNA–SH) in 10 mM PB. Since the clean gold surface is hydrophilic, only a small volume (∼100 µL) of the probe DNA solution was required to completely wet the surface. The 10 µM probe DNA concentration was chosen in order to maximize the surface density as previously reported in the literature.29,33–35,37,38 The gold substrates were incubated in a closed container for approximately 48 hours at room temperature and finally rinsed with PB and dried with nitrogen gas.The DNA-derivatized gold slides were incubated in a 1 mM aqueous solution of 6-mercapto-1-hexanol (MHO) at room temperature for 1 hour in a closed container, rinsed with deionized water and dried with nitrogen gas. Hybridization with the target strands was carried out in 3× hybridization buffer (HB). The slides were covered with a 1 µM solution of target DNA strands in HB for 1 hour at room temperature, rinsed with 3× HB, deionized water and dried in a stream of nitrogen gas. Control experiments included the incubation of bare gold slides in an unmodified DNA solution with the same sequence as the thiol-modified probe and incubation of bare gold slides in MHO solution. At least three samples were prepared for each modification.
Modification of SPR sensing surface with probe DNA
SPR sensing modules were modified by wetting the gold sensor surface with 50 µL of 10 µM DNA–SH solution in PB, and incubating in a closed container for 48 hours at room temperature. The gold surface was rinsed with PB and dried with nitrogen. The DNA–derivatized gold surface was incubated with 100 µL of 1 mM MHO in deionized water and stored for 1 hour in a closed container. The surface was rinsed with deionized water, dried with nitrogen, and then stored in a sealed Petri dish at 4 °C until further use.For the temperature experiment using different surface densities of probe DNA, DNA–SH solutions were prepared in 3× HB: one contained 10 µM DNA–SH and the other contained 10 µM DNA–SH and 10 µM FM target DNA in order to hybridize the probe DNA prior to surface deposition. After 1 hour incubation at room temperature 50 µL of each solution were used to coat the gold surface of four SPR sensors (two for each solution). Following incubation in a closed container for 48 hours at room temperature the gold surface was rinsed with 3× HB and was covered with 100 µL of 1 mM MHO in deionized water. From each set of two sensors incubated in the same DNA–SH solution, one was incubated for 1 hour at room temperature and the other was incubated for 5 hours at room temperature. All surfaces were rinsed with deionized water, dried with nitrogen and stored in a sealed Petri dish at 4 °C until further use.
Electrochemical characterization of gold slides
Electrochemical measurements were obtained with a Gamry Instruments Reference 600 Potentiostat/Galvanostat/ZRA (Warminster, PA, USA). An electrochemical cell consisting of three electrodes was assembled. The modified gold slides were used as the working electrodes, an Accumet saturated calomel electrode (SCE) from Fisher Scientific (Pittsburgh, PA, USA) as the reference electrode, and an Accumet platinum wire as the counter electrode. The slide was contacted with an alligator clip, while an area of 1 cm2 was immersed in the electrolyte solution together with the SCE and counter electrode. The electrolyte solution was prepared in 10 mM PB, containing 1 mM potassium ferricyanide [K3Fe(CN)6] (as redox probe) and 100 mM KCl (as supporting electrolyte).The cyclic voltammetry (CV) curves were obtained in the range of −0.3 V to 0.7 V at a 50 mV s−1 scan rate. The electrochemical impedance spectroscopy (EIS) measurements were performed at a fixed potential of 0.18 V vs.SCE (the reduction potential of ferricyanide ions) and in the frequency range of 0.1 Hz to 100 kHz. The impedance spectra were recorded in the form of Nyquist plots and they were fitted to an equivalent circuit, shown in Fig. 2, based on the Randles and Ershler model.39–41 The modelling was done with Gamry Echem Analyst software, Version 5.10.
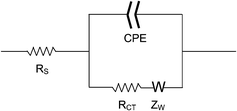 | ||
| Fig. 2 Diagram of the equivalent circuit used to fit the impedance spectroscopy data. | ||
In the presence of a redox probe the model circuit consists of the resistance of the electrolyte solution (RS), the Warburg resistance (ZW) resulting from the diffusion of the redox probe, the charge-transfer resistance (RCT) of the electrode/electrolyte interface on the surface of the working electrode, and a non-ideal capacitance represented by the constant phase element (CPE) due to the non-homogeneity of the interface on the working electrode surface.41–43CPE and RCT represent parameters that are dependent on the solution–electrode interface, thus the nature of the film covering the gold surface. For non-homogeneous films and rough surfaces the impedance data do not fit the theoretical behaviour predicted by the equivalent circuit made of ideally behaving components.40,44 For such cases, the capacitance can be replaced by the constant phase element (CPE)41–43 and any changes in surface coverage are determined by observing changes in the RCT values.33,41,45–49 When molecules with hydrophobic moieties adhere to the gold surface, they form an insulation layer which perturbs the interfacial electron transfer between the electrode and the electroactive species in solution, thereby increasing the charge-transfer resistance. The same effect is observed when charged molecules, such as DNA oligonucleotides, are deposited on the electrode surface. This is attributed to the electrostatic repulsion between the negatively charged DNA layer and the ferricyanide ion, [Fe(CN)6]3−.
Surface plasmon resonance
SPR measurements were performed with an SPR3, High Sensitivity Spreeta Evaluation Kit, from ICx Nomadics. The kit includes parts to assemble a flow cell integrated with the SPR sensor module and an electronic control box to process the signal. The assembly of the flow cell creates three small parallel channels over the gold sensing surface. The dimensions of each of the flow channels are: 9 mm long, 0.8 mm wide and 0.1 mm deep, providing for a 0.72 µL volume per channel. The SPR signal was monitored and recorded using Spreeta 5 software, Version 21.14, from Texas Instruments, Inc. (Dallas, TX, USA). The flow cell was thermally insulated and protected from light by enclosing it in a Styrofoam lined, light-tight box.The sensing area is significantly smaller than the flow channel and is completely covered by the flowing buffer solution during the measurements. The refractive index was monitored while running HB only and then oligonucleotide samples diluted in HB were pumped over the sensor surface. Each SPR measurement was carried out in triplicate by monitoring the SPR response curve for all three channels simultaneously and the average change was reported. The relative amount of target DNA bound to the surface was determined by measuring the net increase of the refractive index over time, compared to that of running buffer alone.
Following hybridization, the sensor surface was regenerated by washing in situ with a 7 M guanidinium chloride aqueous solution for 10 min. Experiments were carried out to determine the effect of the mismatch site on the SPR signal (i.e. 5′, 3′, and middle) at target DNA concentrations ranging from 10 pM to 1 µM. A series of experiments were performed over a range of flow rates (20, 50, 100, 200, 400, and 800 µL min−1) and temperatures (20, 25, 30, 40, 50, and 60 °C). The flow rate was controlled using a Manostat Carter 4/8 multi-channel peristaltic pump from Barnant Co. (Barrington, IL, USA). The temperature was set by immersing the sample tubing in a temperature-controlled water bath before it entered the flow cell. The temperature of running buffer was monitored by inserting a 0.25 mm diameter thermocouple probe, OMEGA Engineering, Inc. (Stamford, CT, USA), inside the lumen of the tube at the closest point possible to the liquid input port on the SPR flow cell. When the temperature of the in-flowing buffer solution reached the desired value the target DNA solution was pumped through the system. For the experiments investigating different flow rates and temperatures the flow time of target DNA was 5 minutes, whereas for the DNA concentration titration the flow time was 30 minutes, since hybridization happens at a slower rate for low DNA concentrations.
Results and discussion
Electrochemical characterization
Cyclic voltammetry is an electrochemical technique that measures the current response of the working electrode to a cyclic linear scan in potential. The presence of an electroactive ion such as [Fe(CN)6]3− in solution allows for charge or electron transfer to occur between the electrode/electrolyte interface due to oxidation or reduction of the redox probe. If the gold surface is unmodified, thus conductive, then two redox current peaks will be observed, whereas no redox current peaks are observed when the gold surface is coated with a film that blocks electron transfer. The cyclic voltammetry (CV) curves of the modified gold electrodes showed no conductivity after the deposition of either thiol-modified (DNA–SH) or non-thiolated (DNA) probe oligonucleotide, but they became conductive again after incubation in 6-mercapto-1-hexanol (MHO) solution (Fig. 3). The lack of redox peaks after modification with DNA–SH or DNA is interpreted as the inability of the [Fe(CN)6]3− ion to penetrate the chemisorbed and physisorbed oligonucleotide film. Physisorption refers to non-specific binding of other functional groups of DNA due to the high surface energy of gold.50 The reappearance of the redox peaks after incubation in MHO solution shows that the redox probe is able to penetrate the mixed monolayer formed on the gold electrode. However, the separation of the redox peaks is larger when compared to those observed for the bare gold indicating an increase in the resistivity of the gold surface, although relatively small, due to the presence of the DNA–SH/MHO mixed monolayer. In support of this view, bare gold slides incubated only in MHO solution for 1 hour provided the same separation of the redox peaks in the CV curve as the DNA–SH/MHO samples (Fig. 3A). The result for the MHO only coated slides is expected as SAMs formed by alkanethiol chains shorter than nine carbons are known not to provide blocking CVs.51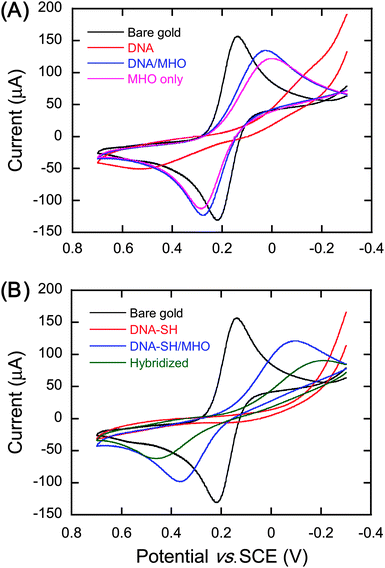 | ||
| Fig. 3 Cyclic voltammograms of bare gold and modified gold slides. (A) Bare gold, gold surface incubated in 10 µM non-thiolated control probe oligonucleotide (DNA), DNA surface incubated in 1 mM 6-mercapto-1-hexanol (MHO) (DNA/MHO), and gold incubated in 1 mM MHO (MHO only). (B) Bare gold, gold surface incubated in 10 µM thiolated probe oligonucleotide (DNA–SH), DNA–SH surface incubated in 1 mM MHO (DNA–SH/MHO), and DNA–SH/MHO surface incubated in 1 µM full match target oligonucleotide (hybridized). | ||
These results appear to show that MHO molecules replace the physisorbed DNA, resulting in the formation of a mixed monolayer of DNA–SH and MHO, which would be suitable for DNA hybridization. After incubation in hybridization buffer with the full match (FM) target strand, the redox peaks of the CV shift outwards (Fig. 3B), showing higher resistivity of the surface towards the [Fe(CN)6]3− ions. This would be expected if the DNA–SH was still bound to the surface so that it can hybridize with the full match target strand. Hybridization of probe DNA results in an increase in the amount of negative charges on the gold surface, thus there would be stronger repulsion towards the negatively charged ferricyanide ions. The increase in resistivity upon surface deposition of DNA oligonucleotides is also observed by impedance spectroscopy, and as it will be discussed below, the CV results are in agreement with the impedance data.
The impedance spectra were recorded in the form of Nyquist plots and the data were fitted to a general equivalent circuit model that contained a CPE parameter instead of capacitance, as shown in Fig. 2. Typical impedance spectra before and after hybridization are shown in Fig. 4. Deposition of negatively charged molecules on the electrode surface results in an increase of the charge-transfer resistance (RCT). Any changes in surface coverage can be followed by observing changes in the RCT values. The charge-transfer resistance of the gold slides after each deposition step is given in Table 2. Slides derivatized with DNA–SH or DNA yielded RCT values at least 100-fold larger than those of bare gold. The high surface energy of plasma cleaned gold increases the affinity of other functional groups in DNA to the surface causing random binding and orientation of the nucleotide strand to the surface.50 Random physisorption of oligonucleotides also results in the wide range of RCT values for both DNA–SH and DNA surfaces (Table 2).
However, the RCT was reduced after the subsequent incubation in MHO solution. As observed in the cyclic voltammetry experiments, this indicates the removal of physisorbed oligonucleotides and the physisorbed portion of DNA–SH oligonucleotides due to the chemisorption of MHO on the gold surface through its thiol group. Incubation in MHO solution provided for consistent RCT values between the samples, while the DNA–SH/MHO surfaces had a larger RCT value compared to the DNA/MHO surfaces, consistent with the expectation that the DNA–SH/MHO surface contains the probe oligonucleotide. In addition the DNA/MHO slides had the same resistance as the MHO monolayer, showing that the DNA layer was completely replaced by MHO. These results further support the view that MHO molecules replace the physisorbed DNA, resulting in the formation of a mixed monolayer of DNA–SH and MHO (Fig. 5).
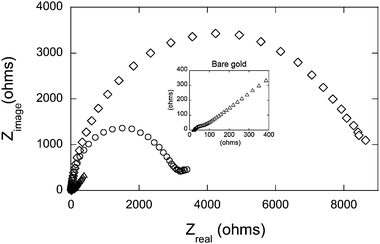 | ||
| Fig. 4 Electrochemical impedance spectra of gold slides recorded in the form of Nyquist plots, prior to surface modification (△ bare gold), after surface modification with the mixed monolayer of thiolated probe and 1 mM 6-mercapto-1-hexanol (○ DNA–SH/MHO), and following hybridization with 1 µM full match target oligonucleotide (◇ DNA–SH/MHO + FM). Impedance measurements were performed in 1 mM K3Fe(CN)6 and 100 mM KCl solution prepared in 10 mM phosphate buffer, pH 7.0. The spectra were obtained in the frequency range from 100 kHz to 0.1 Hz at a fixed DC potential of 0.18 V. Inset: impedance spectrum of the bare gold slide at a smaller scale. | ||
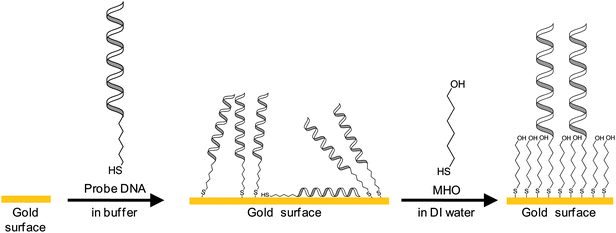 | ||
| Fig. 5 Depiction of the changes on the gold surface after each deposition step. Incubation of gold in the thiolated probe DNA solution coats the gold surface with a mixture of physisorbed and chemisorbed DNA. The CV and impedance results support the view that only after incubation in MHO solution are the probe oligonucleotides available for hybridization due to physisorption and random orientation prior to incubation in MHO solution. | ||
After the hybridization step by incubation in 1 µM full match (FM) target oligonucleotide, the RCT of the DNA–SH/MHO surfaces increased three-fold, whereas the DNA/MHO slides showed no significant change. In addition, hybridization performed on MHO and DNA–SH surfaces (no incubation in MHO solution) did not show any changes in RCT values. The increase in RCT only for the DNA–SH/MHO surfaces strongly supports the idea that the thiolated probe DNA strands are attached to the surface as part of a mixed monolayer with MHO molecules and they are capable of hybridizing with FM target DNA as shown by the increase in resistivity of the surface towards the negatively charged [Fe(CN)6]3− ions.
In a separate experiment, hybridization of DNA–SH/MHO modified slides with each of the four target DNA strands clearly caused a higher resistance than the RCT of the samples exposed to the control target DNA. Exposure to the middle mismatch target strand (P11) yielded the lowest average RCT value compared to the full match strand and the other two one-mismatch strands. In addition, the RCT values for the P11 strand were consistently lower than the RCT of the full match strand. However, the P1 and P21 mismatched target nucleotides did not have significantly different average values from the full match strand (Table 3).
| Target oligonucleotide | Average RCT/kΩ | Standard deviation (n = 9) |
|---|---|---|
| Control | 2.1 | 0.3 |
| FM | 4.9 | 0.7 |
| P11 | 3.1 | 0.8 |
| P1 | 4.0 | 0.5 |
| P21 | 3.9 | 0.5 |
Surface plasmon resonance
The SPR experiments were performed by monitoring the refractive index over time while running the target DNA samples over the sensing surface. Binding of target DNA to the sensor surface is associated with a proportional increase in the refractive index. This change is usually reported in response units (RU) where 1 RU is equivalent to 1 × 10−6 change in the refractive index. When a 1 µM solution of full match (FM) strand was run, the response curve increased by more than 100 RU within one minute and did not return to baseline after rinsing with hybridization buffer (HB) (Fig. 6). Hybridization of the full match strand is shown by the rapid initial linear increase of the response curve. This is due to a higher surface density of binding sites on the sensor surface compared to concentration of target DNA in solution. After the initial rapid increase, the response curve increases slowly, as the binding sites are nearing the saturation point. At first, the slow phase represents the saturation of the binding sites with target DNA strands, whereas afterwards, as the SPR signal slowly rises, it is caused by the accumulation of target DNA on the solution/surface interface as explained by the model of indirect hybridization.52 This theoretical model suggests that prior to hybridization on the surface, the target oligonucleotides must first be non-specifically adsorbed on the surface (i.e. in the solution/surface interface), from where they can diffuse laterally until they find a binding site. According to this model there is equilibrium between oligonucleotides in solution and surface oligonucleotides (non-specifically adsorbed oligonucleotides), which is established only after the binding sites are saturated, as hybridization happens faster than surface adsorption. Therefore, as the binding sites are saturated, the concentration of non-specifically adsorbed DNA on the solution/sensor surface interface slowly increases, resulting in a slow increase of the response curve.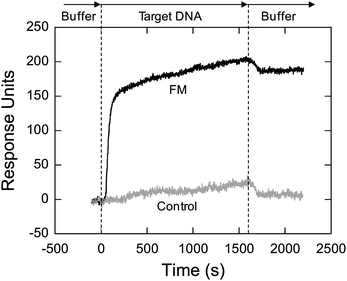 | ||
| Fig. 6 Comparison of SPR response when running solutions of 1 µM non-complementary (control) and 1 µM complementary (FM) target DNA strands over the sensor surface. The SPR experiment was performed in 3× HB, using 100 µL min−1 flow rate at room temperature. Each sensorgram represents the average of three runs. | ||
A 1 µM solution of non-complementary (control) oligonucleotide caused a change in the response curve similar to that of the slow phase of the FM target strand. After switching to buffer, the response curve returned back to the baseline level. Following buffer rinse there is a drop in the response curve for the FM strand too. Therefore, it appears that the slow phase is partially caused by the non-specific adsorption and lateral diffusion of target oligonucleotides on the sensor surface. All RU values reported in this work represent the change in signal with respect to the baseline after the buffer rinse. The sensor surface could be regenerated by washing the surface in situ with a denaturing solution (7 M guanidinium chloride). It was found that the response of the sensor remained the same after at least 20 cycles of hybridization/denaturation steps, allowing for the sensor surface to be reused for subsequent hybridization measurements.
Effect of flow rate and temperature on sensor performance
The rate of analyte delivered to the sensor matrix on the surface, also known as the mass transport of analyte, has been reported to affect the binding or hybridization rate.17,53–55 In order to detect DNA, these SPR systems utilize a 100 nm thick sensing film providing many anchoring sites for attaching probe DNA using biotin–streptavidin. However, for our sensor, the flow rate did not affect the response signal for binding of analyte to the surface over a range of 20–800 µL min−1. This is almost certainly a consequence of the planar, thin (∼2 to 7 nm) sensing film. The binding affinity for DNA hybridization in solution is also known to be affected by temperature.56–58 Based on these references it was expected that at higher temperatures there would be a larger difference in the SPR response of the FM and P11 strands. However, this was not observed up to 50 °C. This indicates that hybridization on the surface is not as sensitive to temperature as hybridization in solution. Nevertheless, at 60 °C there appears to be a slight drop in the signal magnitude for both target strands.It has also been reported that for DNA hybridization on the surface the melting point is affected by the dispersion or availability of the probe DNA on the surface.59 In order to investigate this parameter on our sensor, the effect of temperature on dehybridization was compared at different probes dispersions or surface densities. The dispersion of the probe was varied by using single or double-stranded (hybridized) thiolated probe oligonucleotides to coat the gold surface and incubating surfaces in MHO solution for 1 hour and 5 hours as previously reported.34,35 Ss DNA incubated in MHO for 1 hour is expected to have the highest surface density and double-stranded (ds) DNA incubated in MHO for 5 hours is expected to have the lowest. After running a denaturing solution on each surface, the probes were hybridized with FM strand at room temperature and then the temperature of the running buffer was increased to 60 °C and kept at that temperature for 1 hour. The change in signal magnitude following rehybridization at room temperature varied between the different probe DNA surface densities, indicating that less FM target DNA dehybridized from the surface with higher probe density (Table 4). These results indicate that the apparent melting temperature of DNA hybridized on a surface is compounded by the ability of the probe–target duplex to reorganize itself on the surface. As shown in Table 4, the surface with the highest probe surface density retained most (65%) of the target DNA.
| Probe time in MHO solution | ssDNA 1 hour | ssDNA 5 hours | dsDNA 1 hour | dsDNA 5 hours |
|---|---|---|---|---|
| ΔRU before incubation at 60 °C | 246 | 176 | 197 | 103 |
| ΔRU after incubation at 60 °C | 85 | 103 | 160 | 82 |
| % FM DNA left on the surface | 65% | 41% | 19% | 20% |
Effect of mismatch site on sensor response
A series of hybridization experiments was performed to test the influence of target DNA concentrations for both the FM and the one mismatch strands. As shown in Fig. 7, the signal response was 20% or larger for the hybridization of the FM strand at all concentrations when compared to the P11 strand. The response magnitude of the P21 strand was also lower in magnitude compared to the FM strand at all concentrations. This demonstrates the ability of the sensor to distinguish single nucleotide mismatches in the middle and 3′ position of the target sequence over several orders of magnitude in concentration. The P1 strand showed no significant difference with the FM strand. The smallest concentration measured was 100 pM. Accounting for the baseline noise, the detection limit of the oligonucleotides was determined to be 20 pM, calculated as a triple of baseline noise standard deviations. Assuming a working sample volume of 500 µL and the flow rate to be 20 µL min−1, the detection limit was calculated to be 10 femtomoles.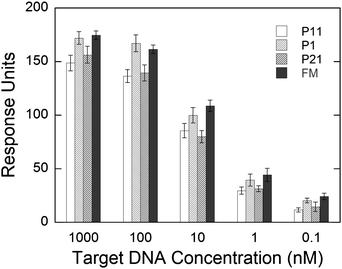 | ||
| Fig. 7 Refractive index change after hybridization of P11, P1, P21, and FM target DNA strands at different concentrations. The hybridization experiments were conducted in 3× HB, using 100 µL min−1 flow rate at room temperature. The differences in RU are statistically significant at all concentrations between the FM and P11 and FM and P21 target strands. Error bars represent the standard deviation between the sensorgrams obtained from the three sensing areas on the SPR module. | ||
The difference in binding between the one-mismatch target strands may be explained in terms of the effects of ionic strength on DNA hybridizations or the proximity of the mismatch to the solution. Following hybridization of the P1 strand the mismatch is formed on the distal end of the duplex or at the DNA monolayer/solution interface, whereas for the P21 strand the mismatch is formed at the proximal end, i.e. closer to the gold surface and away from the oligonucleotide monolayer/solution interface. As both mismatches cause the unpaired ends of the target strands to occupy more space than if they were hybridized, it is the proximal mismatch that is more restrained and feels stronger repulsions from neighboring probe strands, thus reducing the rate of formation of the duplex and its stability. The unpaired base of the P1 strand on the distal end is more flexible and it is more accessible by positive ions and the electrostatic repulsion among DNA strands is reduced by the presence of positive ions. As for the P11 strand it could hybridize with the probe through one half or it can fully hybridize and form a small bulge in the middle of the sequence. The middle position of the mismatch could also allow cross-hybridization between two neighboring probes, as suggested by Levicky and Horgan.60 The P11 strand appears to have the lowest change in magnitude of the refractive index after hybridization thus the smallest number of target DNA oligonucleotides bound to the surface. Therefore, it is more likely that cross-hybridization between two probes is the most common scenario. A depiction of this explanation is shown in Fig. 8.
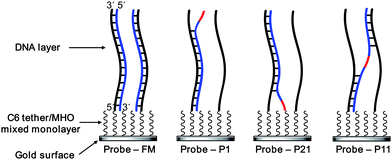 | ||
| Fig. 8 Hybridization of the target DNA strands (blue) to the probe strand (black) on the sensor surface. The mismatched base is shown in red. C6 tether and MHO refer to the hexanethiol tether of the DNA probe and 6-mercapto-1-hexanol, respectively. It is possible for the P11 strand to also bind to one probe DNA, resulting in the formation of a small bulge. | ||
The Langmuir equation shows the dependence of analyte coverage or adsorption on a solid surface from the solution concentration of the analyte in the medium adjacent to the surface. The concentration dependent data were fit to the Langmuir equation (eqn (1)) by performing a non-linear least-squares analysis with Microsoft Excel, as previously described,61 and a modified version of the Langmuir equation was used to fit the experimental data (eqn (2)):62
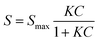 | (1) |
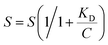 | (2) |
| Target oligonucleotide | Binding affinity (K) (×108 M−1) | Maximum coverage (Smax) |
|---|---|---|
| FM | 2.4 | 169.4 |
| P11 | 1.6 | 146.5 |
| P1 | 2.1 | 169.0 |
| P21 | 1.3 | 153.4 |
Surface coverage of probe DNA
The electrochemistry data demonstrated that incubation of probe DNA coated gold surfaces in MHO solution removed the physisorbed DNA and allowed for spacing of thiolated probe oligonucleotides. The surface densities after incubation in MHO solution have been shown to be in the range of 2.0 × 1012 to 3.8 × 1013 molecules per cm2,34,35 while the highest target hybridization density (7.0 × 1012 molecules per cm2) was obtained after 1 hour incubation in MHO solution.34 As it will be explained below, we believe our probe density is within the range above, which would allow space for efficient hybridization. SPR experiments involving hybridization of target DNA oligonucleotides to probe on the surface have shown that an increase of 1 RU corresponds to 1.2 pg mm−2 surface-bound DNA (regardless of oligonucleotide length).63 For the FM strand (Mw 6486 g mol−1), since the average change in refractive index was 174.7 and each sensing region of the SPR module has a 0.45 mm2 surface area, the average amount of oligonucleotide on the surface after hybridization was calculated to be 7.2 × 10−12 moles per cm2. Dividing by Avogadro's number provides for a target surface density of 4.3 × 1012 molecules per cm2. This means that the density of our probe DNA is equal to or higher than 4.3 × 1012 molecules per cm2. Interestingly, it has been reported that maximum hybridization of probe DNA (with 70–100% hybridization efficiency) is obtained only when the probe DNA density is lower than 4 × 1012 molecules per cm2.34 In addition, surface attachment of a duplexed probe DNA (hybridized prior to immobilization) produced a surface density of 2.8 × 1012 molecules per cm2.35These experimental results are approximately 10-fold smaller than the theoretical maximum coverage of ds DNA based on the diameter of the duplex. Ds DNA has a diameter of 2.4 nm and if oriented perpendicular to the surface, each duplex would cover a circle with a surface area of 4.5 nm2. Assuming maximum surface coverage by hexagonal packing the surface density is calculated to be 2.2 × 1013 molecules per cm2. This implies that steric hindrance is not the only factor that needs to be considered when calculating maximum ds DNA surface coverage. The electrostatic repulsion among the phosphate DNA backbones causes them to require more space than the one calculated by geometric impediments in order for the DNA strands to pack tightly. This is confirmed by the fact that the immobilization of duplexed DNA probe achieved a surface density of only 2.8 × 1012 molecules per cm2, even though it was deposited in 1 M NaCl, which has a relatively high ionic strength and would be able to repress some of the electrostatic repulsion.
Vainrub and Pettitt modelled the effect of electrostatic repulsion forces upon hybridization of 25-base target oligonucleotides and determined that suppression of hybridization begins at surface densities as low as 1012 molecules per cm2.64,65 This could mean that in our sensor surface the hybridized probes are already feeling the presence of neighboring oligonucleotides. In our sensor the spacing of probe DNA appears to be sufficient enough to see a difference in binding between the proximal (3′) and distal (5′) mismatch in the target DNA sequence.
Further calculations were done to determine the average spacing of hybridized probes on our SPR sensor based on the surface density of bound FM target oligonucleotides calculated earlier to be 4.3 × 1012 molecules per cm2. This was done from two different considerations: (1) assuming a random packing of probe DNA on the surface, then the square root of the surface density unit would give the average number of probes per unit length (2.1 × 106 molecules per cm), meaning the average distance or spacing between the hybridized probes would be 4.8 nm. Since duplexed DNA has a diameter of 2.4 nm, then the edge to edge separation between the hybridized probes would be 2.4 nm; (2) assuming ordered packing of the duplexed probes and since their footprint on the surface would be a circle, then they would pack hexagonally and cover only 90.7% of the available surface. Thus a surface density of 4.3 × 1012 molecules per cm2 corresponds to a surface area of each hybridized probe of 2.1 × 10−13 cm2 per molecule, resulting in a distance of 5.2 nm between two hybridized probes. The edge to edge distance would be 2.8 nm.
In both considerations of probe DNA packing, the spacing between hybridized probe DNA strands is theoretically sufficient to fit another probe–target DNA pair, but as both experimental and modelling studies have shown, the sensing surface is already saturated with duplexed DNA. Most likely the overall probe density for our sensor is higher than 4.3 × 1012 molecules per cm2, meaning there are single-stranded probe oligonucleotides between the hybridized ones. And it is the presence of these un-hybridized probes that is probably causing the discrepancy in binding between the one-mismatch target oligonucleotides, depending on the location of the mismatch. Thus, these results indicate that by controlling probe DNA spacing, one could potentially control the ability of the sensor to distinguish between full match and single nucleotide mismatch target, including the magnitude of difference in signal, as a larger (approximately more than 50%) change in signal upon hybridization would be easier to detect.
RNA oligonucleotide detection
The detection of a full match RNA oligonucleotide was also monitored as a function of concentration (Fig. 9). The sensor yielded a very similar SPR response and detection limit as those observed for DNA sensing. These results demonstrate the potential use of this sensing system to rapidly detect short RNA sequences using the DNA probe oligonucleotide. The use of probe RNA oligonucleotides is not practical owing to the instability of RNA compared to DNA.57 This sensor has the potential to be used for detection of RNA directly from biological samples.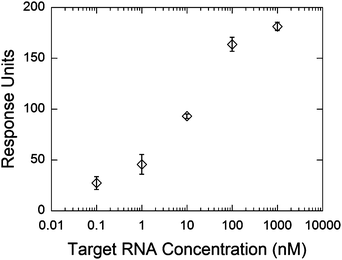 | ||
| Fig. 9 Refractive index change after hybridization of the full complementary RNA strand at different concentrations. The hybridization experiments were conducted in 3× HB, using 100 µL min−1 flow rate at room temperature. Error bars represent the standard deviation between the sensorgrams obtained from the three sensing areas on the SPR module. | ||
Conclusion
In this work, the surface of gold slides and the gold sensing surface of a commercially available SPR sensor were modified by self-assembly to create a mixed monolayer of a thiol modified 21-base long probe DNA and 1-mercapto-6-hexanol (MHO), in order to avoid non-specific adsorption and to facilitate hybridization of target oligonucleotides. The electrochemistry data on the gold slides showed that incubation in MHO solution removed the physisorbed DNA and allowed for spacing of the thiolated probe DNA. No change in the charge transfer resistance (thus, no hybridization) was observed, when DNA–SH or MHO only surfaces were exposed to the FM strand. The SPR sensor was shown to be reproducible over at least 20 hybridization/denaturation cycles. The detection limit was 20 pM or 10 femtomoles, while single nucleotide mismatches could be detected over five orders of magnitude in concentration. RNA was also detected with the same detection limit as DNA. Compared to other SPR-based single mismatch sensors, our system was able to detect mismatches at lower DNA concentrations (100 pM). The site of the mismatch appeared to affect hybridization efficiency. The P11 (middle) and P21 (3′-mismatch) DNA oligonucleotides caused a decrease in the magnitude of the SPR response upon hybridization compared to the FM and P1 (5′-mismatch) oligonucleotides. This shows that single mismatches at the proximal end of the probe result in a less efficient hybridization compared to single mismatches on the distal end of the probe which are closer to the solution.Based on the SPR experiments, it was calculated that the probe DNA density in our sensor is equal to or higher than 4.3 × 1012 molecules per cm2. This value is within the range of reported densities for MHO spaced probe oligonucleotides (2.0 × 1012 to 3.8 × 1013 molecules per cm2) and close to the highest density reported for hybridized target oligonucleotides (7.0 × 1012 molecules per cm2), thus allowing space for efficient target oligonucleotide hybridization. Results indicated that fluctuations in flow rate and temperature should not impede the sensor's ability to detect single nucleotide mismatches. Thus, any fluctuations in these parameters should not affect the read-out of the sensor. This will allow for a practical and applicable sensing system. By using microfluidic platforms, this sensing system could be employed to construct parallel sensing channels for rapid and label-free multiple sample analysis. In addition, this system could potentially be paired with other detection methods, such as impedance spectroscopy, in order to obtain a dual read-out biosensor.
Acknowledgements
This project was partially supported by the US Army Medical Research and Materiel Command (USAMRMC) and the Telemedicine and Advanced Technology Research Center (TATRC) We note that ICx Nomadics (Stillwater, OK, USA) is now ICx Technologies (Arlington, VA, USA).References
- HapMap, Nature, 2003, 426, 789–796 Search PubMed.
- R. A. King, J. I. Rotter and A. G. Motulsky, The Genetic Basis of Common Diseases, Oxford Univ. Press, Oxford, 1992, vol. 20 Search PubMed.
- HapMap, Nature, 2005, 437, 1299–1320 Search PubMed.
- The International HapMap Project, http://www.hapmap.org.
- The Pharmacogenetics and Pharmacogenomics Knowledge Base (PharmGKB), http://www.pharmgkb.org.
- G. Moulin, Rev. Environ. Sci. Bio/Technol., 2005, 24, 101–107 Search PubMed.
- A. Grishok, A. E. Pasquinelli, D. Conte, N. Li, S. Parrish, I. Ha, D. L. Baillie, A. Fire, G. Ruvkun and C. C. Mello, Cell, 2001, 106, 23–34 CrossRef CAS.
- H. W. Hwang and J. T. Mendell, Br. J. Cancer, 2006, 94, 776–780 CrossRef CAS.
- R. C. Lee, R. L. Feinbaum and V. Ambros, Cell, 1993, 75, 843–854 CrossRef CAS.
- K. A. Cissell, S. Shrestha and S. K. Deo, Anal. Chem., 2007, 79, 4754–4761 CAS.
- J. Homola, S. S. Yee and G. Gauglitz, Sens. Actuators, B, 1999, 54, 3–15 CrossRef.
- J. Homola, Anal. Bioanal. Chem., 2003, 377, 528–539 CrossRef CAS.
- K. L. Beattie, W. G. Beattie, L. Meng, S. L. Turner, R. Coral-Vazquez, D. D. Smith, P. M. McIntyre and D. D. Dao, Clin. Chem. (Washington, DC, U. S.), 1995, 41, 700–706 CAS.
- W. Jin, X. Lin, S. Lv, Y. Zhang, Q. Jin and Y. Mu, Biosens. Bioelectron., 2009, 24, 1266–1269 CrossRef CAS.
- J. Ladd, A. D. Taylor, M. Piliarik, J. Homola and S. Jiang, Anal. Chem., 2008, 80, 4231–4236 CrossRef CAS.
- B. P. Nelson, T. E. Grimsrud, M. R. Liles, R. M. Goodman and R. M. Corn, Anal. Chem., 2001, 73, 1–7 CrossRef CAS.
- P. Schuck, Annu. Rev. Biophys. Biomol. Struct., 1997, 26, 541–566 CrossRef CAS.
- E. Kai, S. Sawata, K. Ikebukuro, T. Iida, T. Honda and I. Karube, Anal. Chem., 1999, 71, 796–800 CrossRef CAS.
- B. Persson, K. Stenhag, P. Nilsson, A. Larsson, M. Uhlen and P.-A. Nygren, Anal. Biochem., 1997, 246, 34–44 CrossRef CAS.
- N. Yang, X. Su, V. Tjong and W. Knoll, Biosens. Bioelectron., 2007, 22, 2700–2706 CrossRef CAS.
- S. S. Mark, N. Sandhyarani, C. Zhu, C. Campagnolo and C. A. Batt, Langmuir, 2004, 20, 6808–6817 CrossRef CAS.
- H. Vaisocherova, A. Zitova, M. Lachmanova, J. Stepanek, S. Kralikova, R. Liboska, D. Rejman, I. Rosenberg and J. Homola, Biopolymers, 2006, 82, 394–398 CrossRef CAS.
- L. He, M. D. Musick, S. R. Nicewarner, F. G. Salinas, S. J. Benkovic, M. J. Natan and C. D. Keating, J. Am. Chem. Soc., 2000, 122, 9071–9077 CrossRef CAS.
- A. Nabok, A. Tsargorodskaya, F. Davis and S. P. J. Higson, Biosens. Bioelectron., 2007, 23, 377–383 CrossRef CAS.
- A. Nabok, A. Tsargorodskaya, D. Gauthier, F. Davis, S. P. J. Higson, T. Berzina, L. Cristofolini and M. P. Fontana, J. Phys. Chem. B, 2009, 113, 7897–7902 CrossRef CAS.
- J. Fritz, M. K. Baller, H. P. Lang, H. Rothuizen, P. Vettiger, E. Meyer, H. J. Guntherodt, C. Gerber and J. K. Gimzewski, Science, 2000, 288, 316–318 CrossRef CAS.
- K. Nakatani, A. Kobori, H. Kumasawa and I. Saito, Bioorg. Med. Chem. Lett., 2004, 14, 1105–1108 CrossRef CAS.
- S. Hagihara, H. Kumasawa, Y. Goto, G. Hayashi, A. Kobori, I. Saito and K. Nakatani, Nucleic Acids Res., 2004, 32, 278–286 CrossRef CAS.
- T. Jiang, M. Minunni, P. Wilson, J. Zhang, A. P. F. Turner and M. Mascini, Biosens. Bioelectron., 2005, 20, 1939–1945 CrossRef CAS.
- K. Tawa and W. Knoll, Nucleic Acids Res., 2004, 32, 2372–2377 CrossRef CAS.
- D. Dell'Atti, S. Tombelli, M. Minunni and M. Mascini, Biosens. Bioelectron., 2006, 21, 1876–1879 CrossRef CAS.
- E. Milkani, S. Morais, C. R. Lambert and W. G. McGimpsey, Biosens. Bioelectron., 2010, 25, 1217–1220 CrossRef CAS.
- H. Cai, T. M.-H. Lee and I. M. Hsing, Sens. Actuators, B, 2006, 114, 433–437 CrossRef.
- P. Gong, C.-Y. Lee, J. Gamble Lara, G. Castner David and W. Grainger David, Anal. Chem., 2006, 78, 3326–3334 CrossRef CAS.
- A. W. Peterson, R. J. Heaton and R. M. Georgiadis, Nucleic Acids Res., 2001, 29, 5163–5168 CrossRef CAS.
- E. L. S. Wong, F. J. Mearns and J. J. Gooding, Sens. Actuators, B, 2005, 111–112, 515–521 CrossRef.
- T. M. Herne and M. J. Tarlov, J. Am. Chem. Soc., 1997, 119, 8916–8920 CrossRef CAS.
- T. Ito, K. Hosokawa and M. Maeda, Biosens. Bioelectron., 2007, 22, 1816–1819 CrossRef CAS.
- J. E. B. Randles, Discuss. Faraday Soc., 1947, 1, 11–19 Search PubMed.
- S.-M. Park and J.-S. Yoo, Anal. Chem., 2003, 75, 455A–461A CAS.
- E. Katz and I. Willner, Electroanalysis, 2003, 15, 913–947 CrossRef CAS.
- J. R. Macdonald, Impedance Spectroscopy, Wiley/Interscience, New York, 1987 Search PubMed.
- A. Bardea, E. Katz and I. Willner, Electroanalysis, 2000, 12, 1097–1106 CrossRef CAS.
- D. Savitri and C. K. Mitra, Bioelectrochem. Bioenerg., 1999, 48, 163–169 CrossRef CAS.
- F. Patolsky, A. Lichtenstin and I. Willner, Nat. Biotechnol., 2001, 19, 253–257 CrossRef CAS.
- Y.-T. Long, C.-Z. Li, T. C. Sutherland, H.-B. Kraatz and J. S. Lee, Anal. Chem., 2004, 76, 4059–4065 CrossRef CAS.
- J. Liu, S. Tian, P. E. Nielsen and W. Knoll, Chem. Commun., 2005, 2969–2971 RSC.
- C. Gautier, C. Cougnon, J.-F. Pilard, N. Casse, B. Chenais and M. Laulier, Biosens. Bioelectron., 2007, 22, 2025–2031 CrossRef CAS.
- A. Li, F. Yang, Y. Ma and X. Yang, Biosens. Bioelectron., 2007, 22, 1716–1722 CrossRef CAS.
- A. Paproth, K.-J. Wolter, T. Herzog and T. Zerna, 24th International Spring Seminar on Electronics Technology, Calimanesti-Caciulata, Romania, May 5–9, 2001 Search PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
- D. Erickson, D. Li and J. Krull Ulrich, Anal. Biochem., 2003, 317, 186–200 CrossRef CAS.
- R. Lenigk, R. H. Liu, M. Athavale, Z. Chen, D. Ganser, J. Yang, C. Rauch, Y. Liu, B. Chan, H. Yu, M. Ray, R. Marrero and P. Grodzinski, Anal. Biochem., 2002, 311, 40–49 CrossRef CAS.
- M. Noerholm, H. Bruus, M. H. Jakobsen, P. Telleman and N. B. Ramsing, Lab Chip, 2004, 4, 28–37 RSC.
- P. K. Yuen, G. Li, Y. Bao and U. R. Mueller, Lab Chip, 2003, 3, 46–50 RSC.
- Y. Okahata, M. Kawase, K. Niikura, F. Ohtake, H. Furusawa and Y. Ebara, Anal. Chem., 1998, 70, 1288–1296 CrossRef CAS.
- V. A. Bloomfield, D. M. Crothers and I. Tinoco, Nucleic Acids—Structures, Properties, and Functions, University Science Books, Sausalito, CA, 2000 Search PubMed.
- A. J. Thiel, A. G. Frutos, C. E. Jordan, R. M. Corn and L. M. Smith, Anal. Chem., 1997, 69, 4948–4956 CrossRef CAS.
- J. B. Fiche, A. Buhot, R. Calemczuk and T. Livache, Biophys. J., 2007, 92, 935–946 CAS.
- R. Levicky and A. Horgan, Trends Biotechnol., 2005, 23, 143–149 CrossRef CAS.
- D. C. Harris, J. Chem. Educ., 1998, 75, 119–121 CrossRef CAS.
- M. J. Linman, J. D. Taylor, H. Yu, X. Chen and Q. Cheng, Anal. Chem., 2008, 80, 4007–4013 CrossRef CAS.
- E. Stenberg, B. Persson, H. Roos and C. Urbaniczky, J. Colloid Interface Sci., 1991, 143, 513–526 CrossRef CAS.
- A. Vainrub and B. M. Pettitt, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 66, 041905 CrossRef.
- A. Vainrub and B. M. Pettitt, J. Am. Chem. Soc., 2003, 125, 7798–7799 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |