Optical fiber-based synchronous fluorescence spectroscopy for bacterial discrimination directly from colonies on agar plates
Tourkya
Belal
a,
Karoui
Romdhane
a,
Berdagué
Jean-Louis
b,
Boubellouta
Tahar
a,
Dufour
Eric
a and
Leriche
Françoise
*a
aUPRES 2008.03.0101 Typicité des Produits alimentaires, VetAgro Sup—Campus Agronomique de Clermont, BP 35, 63370, LEMPDES, France. E-mail: f.leriche@vetagro-sup.fr; Fax: +33(0)4 73 98 13 90; Tel: +33(0)4 73 98 13 40
bUR370 Qualité des Produits Animaux, Equipe Typicité Aromatique et Authentification, INRA Centre de Clermont-Ferrand/Theix, F-63122, Saint-Genès-Champanelle, France
First published on 3rd November 2010
Abstract
The development of experimental conditions for rapid bacterial discrimination using fluorescence spectroscopy fingerprinting is presented. Colonies of Pseudomonas and related reference strains on agar plates were analyzed directly using an optic fiber coupled to a laboratory spectrofluorimeter. Spectra were collected using either classic fluorescence spectroscopy after excitation at 250 nm and 340 nm for aromatic amino acids and nucleic acids (AAA + NA) and nicotinamide adenine dinucleotide (NADH) respectively, or synchronous scanning in the excitation wavelength range 250–500 nm. Factorial discriminant analysis (FDA) showed 100% correct classification at the genus and species level from NADH spectra and 100% correct classification at the genus and species level for ∆λ = 30, 70, 90 and 110 nm (cross-validation). Analysis of variance (ANOVA) confirmed that culture time (48 or 72 h) colony and optic fiber positioning had non-significant impacts on differences between species. The use of optical fiber-fluorescence spectroscopy for bacterial discrimination directly on colonies is fast, simple and reliable. The results are independent of culture growth phase and neither need reagent addition nor prior manual preparation of cells, thus eliminating all risk of human error or contamination during sample work-up.
Introduction
It is important to be able to detect, enumerate and identify microorganisms in medical, environmental and food microbiology applications. The qualities required for the analytical methods used depend on their precise objectives, but speed and cost are the most important criteria after the implicit requirements of operators, such as reliability and accuracy, have been met. Despite the development of fast methods, such as the direct epifluorescent filter technique (DEFT), flow cytometry, ATP-based bioluminescence and impedancemetry, enumeration on agar plates using rich or selective culture media remains the principal method used by small and medium-size companies and routine analytical laboratories. This approach is time-consuming and imperfect, since it is only possible to count cultivable microorganisms. Even so, it offers the advantage of a first-line characterization of the flora present by simple macroscopic observation of colony morphology. Due to the lack of selectivity of the culture media, the counted colonies must be identified for a full enumeration result. Most of the commercially available identification systems used routinely by laboratories are based on a series of biochemical tests and substrate utilization. In many cases, and especially for Pseudomonas spp., these methods fail to give an accurate response.9 Numerous PCR-based assays have been developed and are now routinely used in a few reference laboratories. However, alternative tools based on spectroscopic techniques are also being developed, and are attracting increasing interest. Of these, Raman and Fourier transform (FT)-infrared spectroscopy17,19,30–32 are the most fully developed for bacterial discrimination purposes. Other techniques, such as mass spectrometry and related methods including pyrolysis mass spectrometry, gas chromatography-mass spectrometry, and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) have also been successfully used (for a review see ref. 20). Most of them are tedious and need sample preparation and reagents. In the spectroscopy field, fluorescence methods offer several inherent advantages over FT-infrared; for example, they are 100 to 1000 times more sensitive, allowing specific molecular entities to be investigated. Another major advantage of fluorescence is the absence of a water signal.18 Bacteria contain several intrinsic fluorophores that emit photons after excitation in the ultraviolet range.11Tryptophan, phenylalanine and tyrosine are some of the most common fluorescent entities. The nucleotides will also fluoresce, but their quantum yields are about 100 times lower than those of tryptophan.11,27 In addition, several enzymes or cofactors, reduced nicotinamide adenine dinucleotide (NADH) and riboflavin being the most prominent examples, exhibit pronounced native fluorescence after excitation at 320 nm and 380 nm, respectively. The exploitation of this effect was described as early as 1950,42 but it was not until the mid-1980s that the emergence of chemometric methods made it possible to analyze and compare the abundant information obtained from excitation and emission spectra.10,34 Fingerprint spectra generated from whole cells are now considered as good indicators to study bacterial components.6 Several authors have already explored this emerging tool for taxonomic purposes at the genus or species level.1–3,6,12,16,21,26,28,29 For all these studies, bacteria were grown in liquid medium to the exponential growth phase (sample preparation requiring about 36 h) and suspended in saline solution after centrifuging and washing before analysis. A recent paper44 highlights the utility of fluorescence spectroscopy for taxonomic purposes through a standardized procedure independent of the physiological state of bacteria (sample preparation only requiring bacterial suspension and washing from single colonies).The aim of the present study was to improve the method by direct optic fiber reading of colonies on agar plates while conserving or increasing its discriminant potential. The potential of synchronous scanning fluorescence (SyF) spectroscopy was also explored. This technique, largely used in the fields of oil, pharmaceutical, and specific aromatic hydrocarbon analysis, is very useful for the study of mixtures of fluorescent compounds. In the case of whole cells, such as bacteria embedded in complex polysaccharidic matrices, it is probable that numerous intrinsic fluorophores co-occur, with overlapping spectra, causing a loss of information when analyzed at one set excitation wavelength. In SyF spectroscopy both the excitation and emission monochromators are scanned simultaneously in such a manner that a constant wavelength interval is kept between emission and excitation wavelength (∆λ). Using suitable ∆λ and step analysis, SyF reduces spectral overlapping by narrowing spectral bands and simplifying spectra.37Pseudomonas and related collection species were chosen as an exploratory model; “the genus Pseudomonas encompasses arguably the most diverse and ecologically significant group of bacteria on the planet”.43
The manuscript presents the development of the method performed on eleven reference strains and then its application for the discrimination of 37 reference strains. The reference species were selected to cover representatives of phylogenetically unrelated taxa (Burkholderia, Xanthomonas, Burkholderia and Stenotrophomonas), and, within the genus Pseudomonas, relatively unrelated species (Pseudomonas stutzerivs.Pseudomonas chlororaphis) along with related species, e.g.P. chlororaphis, Pseudomonas fragi, Pseudomonas lundensis and Pseudomonas taetrolens. Several biotypes of the same species were also tested (Pseudomonas putida: three strains, Pseudomonas fluorescens: four strains, P. fragi: two strains, P. chlororaphis: two strains, P. stutzeri: two strains and Pseudomonas syringae: two strains).
Materials and methods
Strains and optical fiber analysis
In all, 37 reference type strains were obtained from the Collection of the Institut Pasteur (CIP), Paris, France and from the German Collection of Microorganisms and Cell Cultures (DSM). They are listed in Table 1.| Indicator species and code | Collection ID |
|---|---|
| Pseudomonas | |
| P. fluorescens (P.flu) | CIP 69.13 |
| P. fluorescens biotype B (P.fluB) | CIP 104377 |
| P. fluorescens biotype F (P.fluF) | CIP 59.27 |
| P. fluorescens biotype 3 (P.flu3) | DSM 50124 |
| P. putida (P.put) | CIP 52.191 |
| P. putida biotype A (P.putA) | DSM 50208 |
| P. putida biotype B (P.putB) | DSM 50222 |
| P. fragi (P.frg1) | CIP 55.4 |
| P. fragi (P.frg2) | CIP 60.46 |
| P. chlororaphis (P.chl1) | CIP 63.22 |
| P. chlororaphis (P.chl2) | DSM 50139 |
| P. stutzeri (P.stut1) | CIP 103022 |
| P. stutzeri (P.stut2) | CIP 107689 |
| P. lundensis | CIP 103272 |
| P. taetrolens | CIP 103299 |
| P. pseudoalcaligenes | CIP 66.14 |
| P. synxantha | CIP 59.22 |
| P. resinovorans | CIP 61.9 |
| P. mendocina | CIP 75.21 |
| P. alcaligenes | CIP 101034 |
| P. veronii | CIP 104663 |
| P. monteilii | CIP 104883 |
| P. mosselii | CIP 105259 |
| P. libanensis | CIP 105460 |
| P. migula | CIP 105470 |
| P. orientalis | CIP 105540 |
| P. viridiflava | CIP 106699 |
| P. agarici | CIP 106703 |
| P. cichorii | CIP 106704 |
| P. marginalis | CIP 106712 |
| P. aurantiaca | CIP 109457 |
| P . syringa-syr (P.syr-syr) | DSM 10604 |
| P. syringa-tom (P.syr-tom) | DSM 50315 |
| P. savastanoi | DSM 19341 |
| Xanthomonas | |
| X. campestris | CIP 100069 |
| Stenotrophomonas | |
| S. maltophilia | CIP 60.77 |
| Burkholderia | |
| B. cepacia | CIP 80.24 |
These strains were stored in nutrient broth (Biokar Diagnostics, Beauvais, France) containing 15% glycerol at −80 °C until analyzed. From the pure cultures, bacteria were thawed and inoculated in nutrient broth overnight at 27 °C with shaking, and grown on agar plates with nutrient agar medium (Biokar Diagnostics, Beauvais, France) at 27 °C. To analyze the effect of the culture time (age of the colonies), incubation was performed for 48 h, 72 h and 96 h.
Fluorescence spectroscopy
Fluorescence spectra (FS) and synchronous fluorescence spectra (SyFS) of the bacterial colonies were recorded using a FluoroMax-2 spectrofluorometer (Spex-Jobin Yvon, Longjumeau, France) linked to an optic fiber. The optic fiber was set at 9 mm above the colonies in such a way that the diameter of the light beam was less than 2 mm. The experimental variables were adjusted as follows.First, the optimal conditions of preparation of the bacteria were determined. For this procedure, 11 reference strains were used. They comprised eight species of the genus Pseudomonas: P. stutzeri, P. lundensis, P. taetrolens, P. putida, P. fragi, Pseudomonas pseudoalcaligenes, P. chlororaphis, and P. fluorescens, and three strains of related genera (Pseudomonads): Xanthomonas campestris, Stenotrophomonas maltophilia, and Burkholderia cepacia. Emission fluorescence spectra (280–450 nm, resolution 1 nm, slits 20 nm) were recorded with excitation wavelength set at 250 nm for aromatic amino acids and nucleic acids (AAA + NA). Emission fluorescence spectra (375–550 nm, resolution 1 nm, slits 20 nm) of NADH were also recorded with excitation set at 340 nm. For the each of the two fluorophores and for each strain, four colonies were independently analyzed with two random positions of the optic fiber for each colony. The analysis was performed after three different culture times (ages of colonies): 48 h, 72 h and 96 h of incubation.
Second, the optimal conditions of spectral data acquisition were determined using synchronous fluorescence (SyF) spectroscopy; the excitation wavelength λex and the emission wavelength λem were scanned synchronously with a fixed off-set ∆λ = λem − λex. Synchronous fluorescence spectra (SyFS) were collected in the excitation wavelength range 250–500 nm (resolution 1 nm, slits 18 nm). To find the most suitable wavelength interval (∆λ), scans were recorded first for two strains, P. fragi—CIP 55.4 and P. putida—CIP 52.191, with a ∆λ varying from 20 to 150 nm in 5 nm intervals. Subsequently, ∆λ of 30, 50, 70, 90, 110, and 130 nm were selected for the investigation of 11 strains listed above. Finally, the ∆λ of 30 nm was used to assess the discrimination of the 37 reference strains (Table 1). For each strain, three SyFS spectra were acquired on three distinct colonies with a culture time of 48 h.
Data analysis
To reduce scattering effects and to compare the samples, fluorescence spectra were normalized by reducing the area under each spectrum to a value of 1.47| Xi,j,k,l,n = µ + CTi + Sj + FPk + Cl + εn |
In all, 175 ANOVAs were performed from 264 individual spectra.
In the validation step, the cross-validation method used was leave-one-out cross-validation (LOOCV), where a single observation from the original sample is used as the validation data, and the remaining observations as the training data. This is repeated such that each observation in the sample is used once as validation data.25
The data analyses were processed using the XLStat-pro software (Addinsoft, Paris, France) and the Statistica Software release 6.1 package (Statsoft, Maisons-Alfort, France).
Datasets
For synchronous fluorescence spectra, the number of variables was 250. Data matrices were built from normalized spectra of each ∆λ separately and consisted of 33 observations when the study was performed on 11 strains (11 × 3 spectra per strain) and 111 observations when the study was performed on 37 strains (37 × 3 spectra per strain).
Considering the synchronous fluorescence spectra collected from the 11 strains using the wavelength intervals Δλ of 30, 50, 70, 90, 110, and 130 nm, the FDA was performed separately on the data corresponding to each wavelength interval (Δλ). Thus at the genus and genus-species level, each matrix consisted of 33 observations (11 strains × 3 spectra per strain) and 10 variables (10 PCs). At the species level, each matrix consisted of 24 observations (8 strains × 3 spectra per strain) and 10 variables (10 PCs).
For the synchronous fluorescence spectra collected from the 37 strains using the wavelength interval Δλ of 30 nm, 37 groups were created corresponding to the 37 strains listed in Table 1. Thus the matrix consisted of 111 observations (37 strains × 3 spectra per strain) and 10 variables (10 PCs). We note that three spectra per strain means three spectra collected from three independent colonies of the same strain (collected from the same agar plate).
Results
Method development: intrinsic fluorescence spectra of colony imprint, incidence of optic fiber position and colony culture time
As bacterial colonies are complex evolving mixtures of functional heterogeneous populations embedded in polysaccharide matrices, it was necessary to establish whether UV radiation incidence (optic fiber position) and growth stage had any influence on spectral variance. Thus for each of the 11 reference strains (X. campestris—CIP 100069, S. maltophilia—CIP 6077, B. cepacia—CIP 8024, and Pseudomonas species such as P. stutzeri—CIP 103022, P. lundensis—CIP 103272, P. taetrolens—CIP 103299, P. putida—CIP 52191, P. fragi—CIP 554, and P. pseudoalcaligenes—CIP 26004), the experiment was performed as follows: four independent colonies were analyzed using two random positions of the optic fiber on each colony. The analysis was repeated with three different culture times (48 h, 72 h and 96 h).Fig. 1 presents NADH emission spectra recorded between 375 and 550 nm following excitation at 340 nm of Pseudomonas (fragi), Xanthomonas (campestris), Burkholderia (cepacia) and Stenotrophomonas (maltophilia) colonies (for culture time 48 h). For P. fragi, spectra collected from two optic fiber positions of the same colony and spectra collected from three incubations times (48 h, 72 h and 96 h) are also presented.
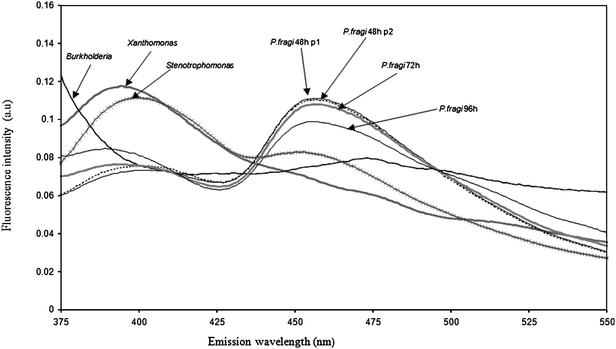 | ||
| Fig. 1 NADH fluorescence spectra recorded after excitation at 340 nm of P. fragiCIP 554 (2 optic fiber positions and 3 culture times), X. campestrisCIP 100069, S. maltophiliaCIP 60.77, and B. cepaciaCIP 80.24 colonies grown on agar plates. | ||
As we see, optic fiber position had no significant effect on the shape of fluorescence spectra collected on the same strain. In addition, no difference could be observed between spectra scanned at 48 h and 72 h of bacterial culture. However, long incubation time seemed to induce some changes to the shape of the spectra. There was also a clear difference between the spectra collected from different genera; marked differences could be observed in the maximum emission band. The positions of the maxima were 456, 473, 395 and 400 nm for Pseudomonas, Burkholderia, Xanthomonas and Stenotrophomonas, respectively. Similar results were observed for the emission spectra collected between 280 and 450 nm after excitation of AAA + NA. The maximum lay between 341 and 369 nm and the shoulders were shifted slightly to lower or higher wavelengths, depending on the bacteria. These results suggest that fluorescence spectra collected directly from bacterial colonies can be considered as fingerprints of bacteria, as had been previously described for bacteria suspensions in liquid medium2,44
No significant variation could be detected between spectra collected for four colonies from the same strain, or from optic fiber positioning on one colony. Even spectral variations observed for the three incubations times of the same strain interfered weakly. However, the species effect was significant, especially for spectral areas located around 417, 456 and 525 nm (Fig. 2).
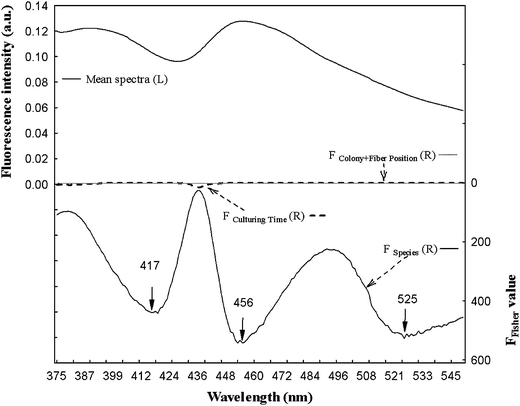 | ||
| Fig. 2 Search by ANOVA for areas of fluorescence spectra significantly influenced by the factors studied. Left scale: Mean spectra of NADH fluorescence spectra and right scale: F of Fisher value for the culture time, species and colony + fiber position factors. | ||
To assess the discrimination potential of the method at the genus and species level, FDA was performed separately on NADH and AAA + NA fluorescence spectral data collected after excitation of the 11 reference strains. The spectra collected after the direct excitation of bacterial colonies were pooled in one matrix (for each fluorophore separately) and this table was analyzed by PCA. Before applying FDA, groups were created as described in Materials and methods.
The confusion matrix resulting from FDA applied to AAA + NA and NADH spectral data collected on colonies at the three incubation times confirmed the excellent grouping of data from the same statistical (taxonomic) cluster. Estimation and cross-validation results are presented in Table 2.
| Fluorophores | Classification level | Index of good classification according to the confusion matrix resulting from FDA | Culturing time (age of colonies) | |||
|---|---|---|---|---|---|---|
| 48 h | 72 h | 96 h | The three CT (48 h + 72 h + 96 h) | |||
| NADH | Genus-species | Cross-validation | 100% | 100% | 96.9% | 90.9% |
| Estimation sample | 100% | 100% | 96.9% | 93.8% | ||
| Genus | Cross-validation | 100% | 97.7% | 100% | 100% | |
| Estimation sample | 100% | 100% | 100% | 100% | ||
| Species | Cross-validation | 100% | 100% | 95.8% | 92.6% | |
| Estimation sample | 100% | 100% | 95.8% | 94.3% | ||
| AAA + NA | Genus-species | Cross-validation | 95.4% | 96.5% | 98.4% | 88.7% |
| Estimation sample | 97.7% | 98.8% | 100% | 92.1% | ||
| Genus | Cross-validation | 95.4% | 97.7% | 95.4% | 97% | |
| Estimation sample | 100% | 100% | 100% | 99.5% | ||
| Species | Cross-validation | 93.7% | 98.4% | 95.8% | 90.3% | |
| Estimation sample | 98.4% | 100% | 100% | 93.7% | ||
Considering cross-validation analysis, the percentage of correct classification varied from 93.7% (AAA + NA excitation, culture time 48 h/species discrimination) to 100% (NADH excitation, culture time 48 h or 72 h/genus or species discrimination). For the estimation sample, correct positioning could be observed with a percentage varying from 95.8% (NADH excitation, 96 h, colonies/species discrimination) to 100% (NADH excitation, 48 h or 72 h, colonies/genus or species discrimination). The results indicated that 96 h of culture induced a few errors in genera or species classification, as suspected by the ANOVA results. Finally, 100% correct classification at the genus and species level was obtained for NADH fluorophore after 48 h culture on agar plates.
Method development: synchronous fluorescence spectra (SyFS)
SyF spectra collected at ∆λ 30, 50, 70, 90, 110, and 130 nm for P. fragiCIP 55.4 aged 48 h are presented Fig. 3.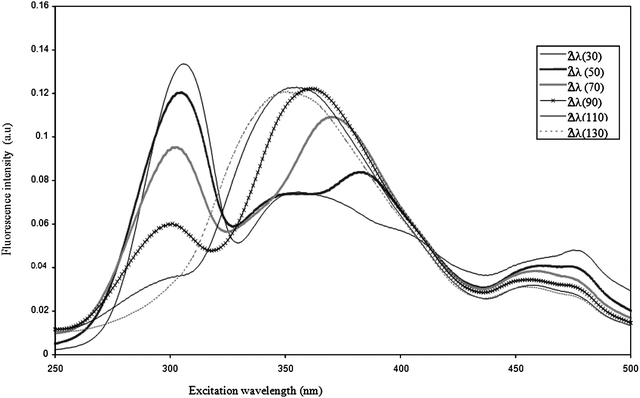 | ||
| Fig. 3 Excitation synchronous fluorescence spectra of P. fragiCIP 55.4 at ∆λ 30, 50, 70, 90, 110 and 130 nm. | ||
SyFs present marked differences with regard to the Δλ which defines overlapping of absorption and emission bands. For the P. fragi strain, the spectra of which are presented in Fig. 3, the first principal band was very intense at Δλ 30 nm, but decreased markedly as Δλ increased, totally disappearing at Δλ 130 nm. By contrast, the small shoulder observed at about 360 nm for Δλ 30 nm became the sole band at Δλ 130 nm. Hence each Δλ provides specific information related to the fluorophore or mixture of fluorophores analyzed. Similar results were observed for all the strains studied.
To determine the best wavelength interval (∆λ) that allowed the best discrimination of bacterial strains, FDA was performed on spectral data corresponding to each ∆λ for the 11 strains.
The discrimination potential of the method at the genus level was evaluated by creating four groups before applying FDA as described for NADH and AAA + NA spectra. These were the Pseudomonasgroup containing the eight reference species, the Xanthomonasgroup (X. campestris), the Stenotrophomonasgroup (S. maltophilia) and the Burkholderiagroup (B. cepacia).
Similarly, to assess the discrimination power of the method at the species level, eight groups were created close to each Pseudomonas species considered (stutzeri, lundensis, teatrolens, putida, fragi, chlororaphis, pseudoalcaligenes, and fluorescens). The results are presented in Table 3.
| Intervals wavelength (∆λ) | Index of good classification according to the confusion matrix resulting from FDA | Classification level | ||
|---|---|---|---|---|
| Genus | Species | Genus-species | ||
| ∆λ (30) | Cross-validation | 100% | 100% | 100% |
| Estimation sample | 100% | 100% | 100% | |
| ∆λ (50) | Cross-validation | 100% | 95.8% | 100% |
| Estimation sample | 100% | 100% | 100% | |
| ∆λ (70) | Cross-validation | 96.9% | 100% | 100% |
| Estimation sample | 100% | 100% | 100% | |
| ∆λ (90) | Cross-validation | 100% | 100% | 96.9% |
| Estimation sample | 100% | 100% | 100% | |
| ∆λ (110) | Cross-validation | 93.9% | 100% | 96.9% |
| Estimation sample | 100% | 100% | 100% | |
| ∆λ (130) | Cross-validation | 93.9% | 95.8% | 96.9% |
| Estimation sample | 100% | 100% | 100% | |
Whatever the ∆λ chosen for bacterial analysis, 100% correct classification was observed from the four genera and the eight species.
Concerning cross-validation analysis, results varied from 96.9% (∆λ 90, 110 and 130 nm) to 100% for ∆λ 30, 50 and 70 nm at the genus-species level. Nevertheless, as minor errors could be observed for ∆λ of 50 nm (cross-validation at the species level) and for ∆λ of 70 nm (cross-validation at the genus level), the wavelength interval of 30 nm was considered in the last part of the study.
Application of the valid method for the discrimination of 37 reference strains
The 37 reference strains presented in Table 1 were finally analyzed according to the following protocol: each bacterial strain was grown in triplicate on solid agar plates for 48 h. Three synchronous spectra were collected for each colony in the 250–500 nm range with a constant wavelength difference of 30 nm.Collected data were pooled in one matrix and PCA was applied. The first 10 PCs were used as new variables to perform FDA. Before applying FDA, 37 groups were created corresponding to the 37 strains used.
On the factorial map shown in Fig. 4, F1 and F2 factors represented only 62.7% of the total variance. However, they allowed correct discrimination of most of the analyzed strains. Only, P. stutzeri1, P. lundensis, P. taetrolens, P. putida, P. fragi 1, P. fluorescens, P. fragi 2, Pseudomonas mentelii, Pseudomonas orientalis, Pseudomonas cichorii, Pseudomonas marginalis, P. stutzeri2 and P. putida B formed a compact group that was not differentiated by the 2D projection alone. According to the F3 discriminant factor, which accounted for 11.8% of the total variance, these strains were well separated (data not shown).
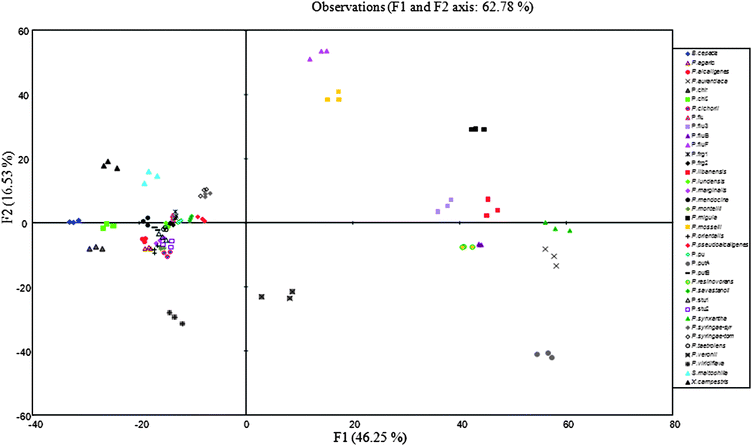 | ||
| Fig. 4 Discriminant analysis similarity map determined by discriminant factors 1 and 2 for the synchronous fluorescence (∆λ 30 nm) spectral data of Pseudomonas reference type strains. | ||
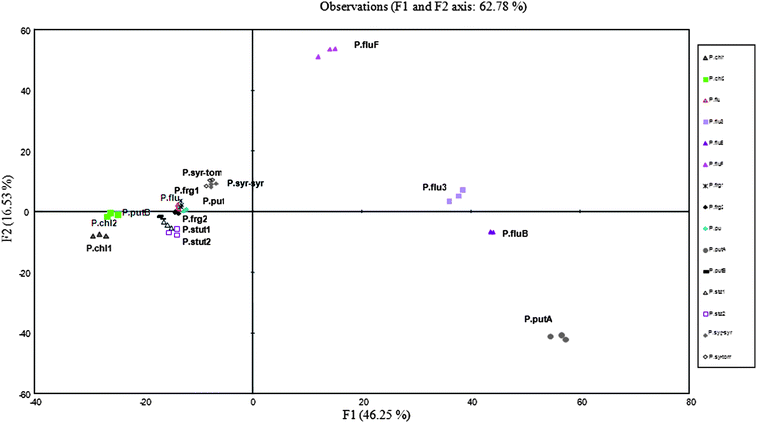
Let us now consider more precisely the species for which several biovars or biotypes were included in our study. For P. fluorescens, they were P. fluorescens CIP 69.13 (P.flu), P. fluorescens biotype B CIP 104377 (P.fluB), P. fluorescens biotype F CIP 59.27 (P.fluF) and P. fluorescens biotype III DSM 50124 (P.flu3). For P. putida, they were P. putida CIP 52.191(P. put), P.putida biotype A DSM 50208 (P.putA) and P. putida biotype B DSM 50222 (P.putB). For P. fragi they were P. fragi CIP 55.4 (P.frg1) and P. fragi CIP 60.46 (P.frg2). For P. chlororaphis, they were P. chlororaphis CIP 63.22 (P.chl1) and P. chlororaphis DSM 50139 (P.chl2). For P. stutzeri they were P. stutzeri CIP 103022 (P.stut1), P. stutzeri CIP 107689 (P.stut2) and for P. syringae, they were P. syringae pvar syringae DSM 10604 (P.syr-syr) and P. syringae pvar tomato DSM 50315 (P.syr-tom). Fig. 5 presents their positioning on the factorial discriminant map resulting from FDA.
 | ||
| Fig. 5 Discriminant analysis similarity map determined by discriminant factors 1 and 2 for the synchronous fluorescence (Δλ 30 nm) spectral data of 37 Pseudomonas reference strains; focus on the species P. fluorescens, P. putida, P. fragi, P. chlororaphis, P. syringae, P. stutzeri and their biotypes or biovars. | ||
Albeit on a small number of strains, our results confirm the taxonomic homogeneity within the P. fragi, P. chlororaphis, P. stutzeri or P. syringae species, spectral data from the same species being very closely positioned on the factorial map (P.frg1 and P.frg2, P.chl1 and P.chl2, P.stut1 and P.stut2, P.syr-syr and P.syr-tom).
Concerning P. fluorescens, spectral data analysis was particularly interesting. In particular, according to discriminant factor F1, which accounted for 45.2% of the total variance, P.flu was well separated from P.fluB, P.fluF and P.flu3. These latter were well separated according to factor F2, which accounted for 16.5% of the total variance. However, while P.flub and P.flu3 presented high relatedness, P.flu appeared more closely related to P.frag1 and P.frag2.
The three biovars of P. putida (P.put, P.putA, P.putB) were well separated on the F1 axis. P.putB and P.put presented close relatedness, but were clearly distant from P.putA, which was positioned close to P.fluB, P.fluIII and P.flu F.
In Fig. 6a are presented the average spectra of the 3 strains of P. putida: P.put, P.putB, P.putA collected according to the protocol described previously. This figure makes it possible to clearly visualize heterogeneity many time described for P. putida. The spectrum of the P.putA strain is distinguished clearly from both others by a peak of strong intensity in the area of 414 nm. For comparison, the spectra of the 4 strains of P. fluorescens are presented in Fig. 6b. There still, the strains heterogeneity is visualized. The taxonomic proximity between the P.putA strains and the P.flub and P.fluIII strains of P. fluorescens finds its explanation in the presence of the same peak in the area of 415–417 nm.
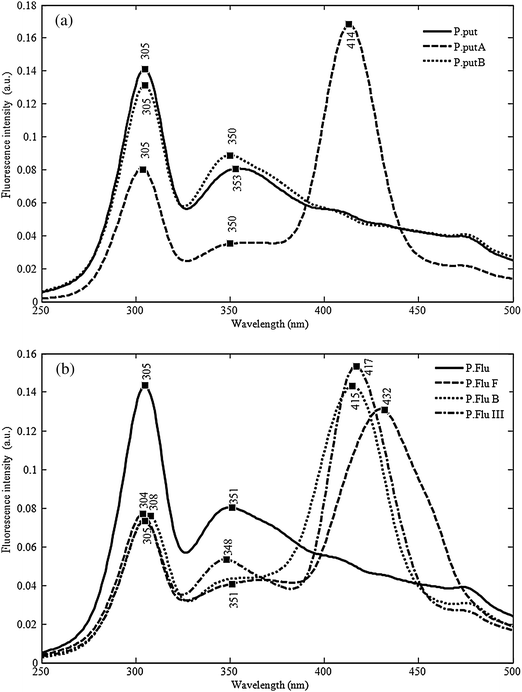 | ||
| Fig. 6 (a) Synchronous fluorescence spectra of three P. putida biotypes recorded in the 260–500 nm range with a constant wavelength difference of 30 nm. (b) Synchronous fluorescence spectra of four P. fluorescens biotypes recorded in the 260–500 nm range with a constant wavelength difference of 30 nm. | ||
The current state of the knowledge does not allow the identification of the molecule responsible so the effect remains unexplained.
These results highlight the strong discriminating power of the method and also confirm the heterogeneity of these two last species as observed by numerous authors.4,48,49
Overall, confusion matrix analysis from FDA presented 97.3% correct classifications for the estimation sample and 94.5% correct classification for the cross-validation (data not shown). Among the 111 collected spectra, only four misclassifications were detected; they concerned confusion between P. marginalis (one spectrum classified as P. lundensis), P. putida (one spectrum classified as P. taetrolens), Pseudomonasviridiflava (one spectrum classified as Pseudomonas syringae-syr), and P. fluorescens (one spectrum classified as Pseudomonas monteilii). Finally, the discriminant ability of the method is confirmed since the grouping of the strains from the same species was obtained and separation of strains from different species and even from different biotypes of the same strain was observed.
Discussion
Microbial cells retain numerous biological molecules such as amino acids, tryptophan, tyrosine, phenylalanine, enzymes and coenzymes FAD, flavins, NADH or NADPH which fluoresce when excited at specific wavelengths. Excitation and/or emission spectra characterize the environment of the fluorophore and subtle differences between spectra can be analyzed using appropriate statistical methods. Since the middle of the last decade, several authors have explored this phenomenon and demonstrated that fluorescence spectra collected from bacterial cells could be considered as fingerprints of the bacteria, making it possible to discriminate between microorganisms belonging to different taxonomic families,6,33 or bacteria at the family, genus, species, and the subspecies level.2 Accurate discrimination (0% error) between Lactococcus lactis, Pediococcus pentosaceus, Kocuria varians, P. fluorescens and Listeria innocua was observed by Ammor et al.3 using tryptophan spectra. These authors also successfully utilized fluorescence properties of the three aromatic amino acids (tryptophan, tyrosine and phenylalanine) and nucleic acids (DNA and RNA) (AAA + NA) to discriminate lactic acid bacteria (LAB) and human bifidobacteria at the genus, species and subspecies level (97% correct classification). In a similar way, Kunnil obtained excellent clustering of Bacillus species isolated or as part of dust aerosols. For all of these studies, fluorescence analyses were performed on microbial liquid cultures pseudo-synchronized in exponential phase or on bacterial suspensions after elimination of culture medium by centrifuging and washing.The main objective of our work was to evaluate the possibility of direct analysis of bacterial colonies on solid agar medium. Two obstacles were considered: (i) many Pseudomonas spp. produce siderophores,35 which are highly fluorescent molecules, so intensive peaks could appear in collected spectra, masking minor modifications between species and (ii) bacterial colonies are highly heterogeneous populations in terms of physiologic state, and genotypic and phenotypic status.13 In addition, the complex matrices that enclose bacterial cells retain numerous metabolites produced during bacterial growth, so that heterogeneity within colonies could be expected to hinder analytical reproducibility. In a previous study,44 we reported the fluorescence spectroscopy analysis of the same 11 reference strains presented in the first part of this work. Bacteria were collected from colonies and washed twice before being suspended in saline solution, so that all secreted metabolites were eliminated by washing. In these conditions, more than 90% accurate discrimination of Pseudomonas at the genus and species level had been observed. The discrimination score reached 97.9% when AAA + NA fluorophores were used for species analysis. In the present study, direct analysis of Pseudomonas colonies gave similar results for AAA + NA fluorophores. Better results were obtained using NADH fluorophores, the percentage of discrimination at the genus and species level reaching 100%. This result is surprising since NADH fluorescence intensity is known to be closely related to the metabolic activity of the cell and thus to its physiological age and growth conditions. We can assume that spectra collected from colonies are averaged spectra of each excited cell, erasing cell-to-cell variations.
To our knowledge, SyFS has not yet been assessed for bacteria discrimination. Our result highlights the power of this technique for Pseudomonad characterization, 100% correct classification of the strains at the genus, species and subspecies level being obtained.
The positioning of the fiber was found not to have any influence on the spectra collected from the same colony, and negligible variations were detected when spectra were collected from 48 h or 72 h of culture on solid agar medium. The use of optical fiber-fluorescence spectroscopy for bacterial discrimination directly from colonies is fast and simple. The results are independent of culture growth phase, and they are achieved without addition of reagents or manual preparation of cells, eliminating all human error or contamination during the preparation of the samples.
Our results clearly demonstrate the reproducibility and reliability of the method. We can therefore consider that from a practical point of view the method is valid in the described conditions for Pseudomonas discrimination: SyFS scanning at a constant wavelength interval of 30 nm of bacterial colonies after 48 h to 72 h culture.
More largely, this radically new approach (along with other spectroscopic methods) should supply new information for Pseudomonads taxonomy. This group, which is characterized by its broad phenotypic and genotypic diversity, has been the subject of extensive taxonomic studies for several decades.43 However, despite the implementation of molecular methods based on DNA–DNA hybridization experiments,15 16S or 23S rRNA sequencing4 or the use of specific markers such as oprI,14 gyrB, rpoD,46atpD, carA, recA22 or oprF,7 delineation of Pseudomonads has been considerably improved but remains imperfect, with species poorly described including even well-known species such as P. putida and P. fluorescens. In this context, methods based on chemotaxonomic marker analysis offer a new utility. Some of them concern the analysis of particular cell components such as cellular fatty acid methyl esters,40 whole-cell protein45 or siderophores.36 For the same reason, spectroscopic methods have become a growing field of interest. Among these, mass spectroscopy39 and infrared spectroscopy8 have been well described. Fluorescence spectroscopy coupled with appropriate chemometric treatment of data has come to be considered as a powerful and fast tool of strain proximity for phylogenetic analysis as pointed out by Peix and colleagues in a recent review.38
The present study now opens the door to more applications such as the identification of a mixture of Pseudomonads. This is already in process in our laboratory and the results are very encouraging but a large spectrum library has still to be built containing spectra for reference and well-identified strains. In these conditions, it is possible to consider the ultra fast identification of unknown isolates starting from a heterogenic mixture of colonies on agar plates. So, for example, more than 48 hours would be gained by setting up an analytical method of reference like ISO/TS 11059:2009.23
Acknowledgements
We thank Barabra Roupioz for technical assistance. This work was supported by a research grant from the Albaath University of Syria.References
- A. Alimova, A. Katz, H. E. Savage, M. Shah, G. Minko, D. V. Will, R. B. Rosen, S. A. McCormick and R. R. Alfano, Native fluorescence and excitation spectroscopic changes in Bacillus subtilis and Staphylococcus aureus bacteria subjected to conditions of starvation, Appl. Opt., 2003, 42(19), 4080–4087 CrossRef CAS.
- M. Ammor, Recent advances in the use of intrinsic fluorescence for bacterial identification and characterization, J. Fluoresc., 2007, 17(5), 455–459 CrossRef CAS.
- S. Ammor, K. Yaakoubi, I. Chevallier and E. Dufour, Identification by fluorescence spectroscopy of lactic acid bacteria isolated from a small-scale facility producing traditional dry sausages, J. Microbiol. Methods, 2004, 59(2), 271–281 CrossRef CAS.
- Y. Anzai, H. Kim, J. Y. Park, H. Wakabayashi and H. Oyaizu, Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence, Int. J. Syst. Evol. Microbiol., 2000, 50(4), 1563–1589 Search PubMed.
- D. Bertrand, P. Courcoux and E. M. Qannari, Méthodes exploratoires, in La spectroscopie infrarouge et ses applications analytiques, ed. D. Bertrand and E. Dufour, Lavoisier, Paris, 2006, pp. 317–345 Search PubMed.
- H. Bhatta, E. M. Goldys and R. P. Learmonth, Use of fluorescence spectroscopy to differentiate yeast and bacterial cells, Appl. Microbiol. Biotechnol., 2006, 71(1), 121–126 CrossRef CAS.
- J. Bodilis and S. Barray, Molecular evolution of the major outer-membrane protein gene (oprF) of Pseudomonas, Microbiology, 2006, 152(4), 1075–1088 CrossRef CAS.
- A. Bosch, A. Minan, C. Vescina, J. Degrossi, B. Gatti, P. Montanaro, M. Messina, M. Franco, C. Vay, J. Schmitt, D. Naumann and O. YantornoFourier Transform Infrared Spectroscopy for Rapid Identification of Nonfermenting Gram-Negative Bacteria Isolated from Sputum Samples from Cystic Fibrosis Patients, J. Clin. Microbiol., 2008, 46, 2535–2546 Search PubMed.
- P. P. Bosshard, R. Zbinden, S. Abels, B. Boddinghaus, M. Altwegg and E. C. Bottger, 16S rRNA gene sequencing versus the API 20 NE system and the VITEK 2 ID-GNB card for identification of nonfermenting Gram-negative bacteria in the clinical laboratory, J. Clin. Microbiol., 2006, 44(4), 1359–1366 CrossRef CAS.
- S. K. Brahma, M. P. Baek, D. Gaskill, R. K. Force, W. H. Nelson and J. Sperry, The rapid identification of bacteria using time-resolved fluorescence and fluorescence excitation spectral methods, Appl. Spectrosc., 1985, 39, 869–872 CrossRef CAS.
- C. R. Cantor and P. R. Schimmel, Other Optical Techniques. Biophysical Chemistry-Part 2: Techniques for the Study of Biological Structure and Function, W. H. Freeman, New York, 1980, pp. 409–480 Search PubMed.
- W. Charlotte, Comparison of chemometric methods for classification of fungal extracts based on rapid fluorescence spectroscopy, J. Chemom., 2000, 14(5–6), 765–776 CrossRef CAS.
- L. P. Choo-Smith, K. Maquelin, T. van Vreeswijk, H. A. Bruining, G. J. Puppels, N. A. N. Thi, C. Kirschner, D. Naumann, D. Ami, A. M. Villa, F. Orsini, S. M. Doglia, H. Lamfarraj, G. D. Sockalingum, M. Manfait, P. Allouch and H. P. Endtz, Investigating microbial (micro)colony heterogeneity by vibrational spectroscopy, Appl. Environ. Microbiol., 2001, 67(4), 1461–1469 CrossRef CAS.
- D. De Vos, C. Bouton, A. Sarniguet, P. De Vos, M. Vauterin and P. Cornelis, Sequence diversity of the oprI gene, coding for major outer membrane lipoprotein I, among rRNA group I Pseudomonads, J. Bacteriol., 1998, 180(24), 6551–6556 CAS.
- P. De Vos, A. van Landschoot and P. Segers, et al. Genotypic relationships and taxonomic localization of unclassified Pseudomonas and Pseudomonas-like strains by deoxyribonucleic acid: ribosomal ribonucleic acid hybridizations, Int. J. Syst. Bacteriol., 1989, 39, 35–49 Search PubMed.
- S. Determann, J. M. Lobbes, R. Reuter and J. Rullkotter, Ultraviolet fluorescence excitation and emission spectroscopy of marine algae and bacteria, Mar. Chem., 1998, 62(1), 137–156 CrossRef CAS.
- B. Dziuba, A. Babuchowski, D. Nalecz and M. Niklewicz, Identification of lactic acid bacteria using FTIR spectroscopy and cluster analysis, Int. Dairy J., 2007, 17(3), 183–189 CrossRef.
- C. Estes, A. Duncan, B. Wade, C. Lloyd, W. Ellis and L. Powers, Reagentless detection of microorganisms by intrinsic fluorescence, Biosens. Bioelectron., 2003, 18(5–6), 511–519 CrossRef CAS.
- Z. Filip and S. Hermann, An attempt to differentiate Pseudomonas spp. and other soil bacteria by FT-IR spectroscopy, Eur. J. Soil Biol., 2001, 37, 137–143 CrossRef CAS.
- A. Fox, Mass spectrometry for species or strain identification after culture or without culture: past, present, and future, J. Clin. Microbiol., 2006, 44(8), 2677–2680 CrossRef CAS.
- H. I. Heaton, Principal-components analysis of fluorescence cross-section spectra from pathogenic and simulant bacteria, Appl. Opt., 2005, 44(30), 6486–6495 CrossRef.
- E. Hilario, T. R. Buckley and J. M. Young, Improved resolution on the phylogenetic relationships among Pseudomonas by the combined analysis of atp D, car A, rec A and 16S rDNA, Antonie van Leeuwenhoek, 2004, 86(1), 51–64 CrossRef CAS.
- International Organization for Standardization, ISO/TS 11059:2009: Milk and milk products—Method for the Enumeration of Pseudomonas spp, International Organization for Standardization, Geneva, Switzerland, 2009 Search PubMed.
- I. T. Jolliffe, Principal Component Analysis, Springer, New York, 1986 Search PubMed.
- R. Kohavi, A Study of Cross Validation and Bootstrap for Accuracy Estimation and Model Selection. Proceeding of the Fourteenth International Joint Conference on Artificial Intelligence, vol. 2(12), 1995, pp. 1137–1143 Search PubMed.
- J. Kunnil, Identification Studies of Bacillus Spores Using Fluorescence Spectroscopy, Department of Physics and Astronomy, University of Canterbury, Christchurch, New Zealand, 2005, p. 174 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum, New York, 2nd edn, 1999 Search PubMed.
- L. Leblanc and E. Dufour, Monitoring the identity of bacteria using their intrinsic fluorescence, FEMS Microbiol. Lett., 2002, 211(2), 147–153 CrossRef CAS.
- F. Leriche, A. Bordessoules, K. Fayolle, R. Karoui, K. Laval, L. Leblanc and E. Dufour, Alteration of raw-milk cheese by Pseudomonas spp.: monitoring the sources of contamination using fluorescence spectroscopy and metabolic profiling, J. Microbiol. Methods, 2004, 59(1), 33–41 CrossRef CAS.
- K. Maquelin, L. P. Choo-Smith, H. P. Endtz, H. A. Bruining and G. J. Puppels, Rapid identification of Candida species by confocal Raman microspectroscopy, J. Clin. Microbiol., 2002, 40(2), 594–600 CrossRef CAS.
- K. Maquelin, C. Kirschner, L. P. Choo-Smith, N. A. Ngo-Thi, T. van Vreeswijk, M. Stammler, H. P. Endtz, H. A. Bruining, D. Naumann and G. J. Puppels, Prospective study of the performance of vibrational spectroscopies for rapid identification of bacterial and fungal pathogens recovered from blood cultures, J. Clin. Microbiol., 2003, 41(1), 324–329 CrossRef CAS.
- L. Mariey, J. P. Signolle, C. Amiel and J. Travert, Discrimination, classification, identification of microorganisms using FTIR spectroscopy and chemometrics, Vib. Spectrosc., 2001, 26(2), 151–159 CrossRef CAS.
- H. Y. Mason, C. Lloyd, M. Dice, R. Sinclair, W. Ellis and L. Powers, Taxonomic identification of microorganisms by capture and intrinsic fluorescence detection, Biosens. Bioelectron., 2003, 18, 521–527 CrossRef CAS.
- R. R. Meglen, Chemometrics: its role in chemistry and measurement sciences, Chemom. Intell. Lab. Syst., 1988, 3(1–2), 17–29 CrossRef CAS.
- J.-M. Meyer, Pyoverdines: pigments, siderophores and potential taxonomic markers of fluorescent Pseudomonas species, Arch. Microbiol., 2000, 174(3), 135–142 CrossRef CAS.
- J.-M. Meyer, C. Gruffaz, V. Raharinosy, I. Bezverbnaya, M. Schäfer and H. Budzikiewicz, Siderotyping of fluorescent Pseudomonas: molecular mass determination by mass spectrometry as a powerful pyoverdine siderotyping method, BioMetals, 2008, 21(3), 259–271 Search PubMed.
- D. Patra and A. K. Mishra, Recent developments in multi-component synchronous fluorescence scan analysis, TrAC, Trends Anal. Chem., 2002, 21(12), 787–798 CrossRef CAS.
- A. Peix, M.-H. Ramírez-Bahena and E. Velázquez, Historical evolution and current status of the taxonomy of genus Pseudomonas, Infect., Genet. Evol., 2009, 9, 1132–1147 CrossRef.
- E. Postnikova, C. Baldwin, C. A. Whitehouse, A. Sechler, N. W. Schaad, R. Sampath, V. Harpin, F. Li, R. Melton, L. Blyn, J. Drader, S. Hofstadler and W. L. Schneider, Identification of bacterial plant pathogens using multilocus polymerase chain reaction/electrospray ionization-mass spectrometry, Phytopathology, 2008, 98(11), 1156–1164 CrossRef.
- M. Quezada, G. Buitron, I. Moreno-Andrade, G. Moreno and L. M. Lopez-Marin, The use of fatty acid methyl esters as biomarkers to determine aerobic, facultatively aerobic and anaerobic communities in wastewater treatment systems, FEMS Microbiol. Lett., 2007, 266(1), 75–82 CrossRef CAS.
- M. Safar, D. Bertrand, P. Robert, M. Devaux and C. Genot, Characterization of edible oils, butters and margarines by Fourier transform infrared spectroscopy with attenuated total reflectance, J. Am. Oil Chem. Soc., 1994, 71(4), 371–377 CrossRef CAS.
- S. Sambursky and G. Wolfsohn, Measurement of fluorescence spectra, J. Opt. Soc. Am., 1948, 38(8), 739 CrossRef CAS.
- A. J. Spiers, A. Buckling and P. B. Rainey, The causes of Pseudomonas diversity, Microbiology, 2000, 146(10), 2345–2350 CAS.
- B. Tourkya, T. Boubellouta, E. Dufour and F. Leriche, Fluorescence spectroscopy as a promising tool for a polyphasic approach to Pseudomonad taxonomy, Curr. Microbiol., 2009, 58(1), 39–46 CrossRef CAS.
- D. M. Weller, Pseudomonas biocontrol agents of Soilborne pathogens: looking back over 30 years, Phytopathology, 2007, 97(2), 250–256 CrossRef.
- S. Yamamoto, H. Kasai, D. L. Arnold, R. W. Jackson, A. Vivian and S. Harayama, Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes, Microbiology, 2000, 146(10), 2385–2394 CAS.
- D. Bertrand and C. N. G. Scotter, Application of Multivariate Analyses to NIR Spectra of Gelatinized Startch, Appl. Spectrosc., 1992, 46, 1420–1425 CrossRef CAS.
- L. Ait Tayeb, E. Ageron, F. Grimont and P. A. D. Grimont, Molecular phylogeny of the genus Pseudomonas based on rpoB sequences and application for the identification of isolates, Res. Microbiol., 2005, 156, 763–773 CrossRef CAS.
- Bossis, P. Lemanceau, X. Latour and L. Gardan, The taxonomy of Pseudomonas fluorescens and Pseudomonas putida: current status and need for revision, Agronomie, 2000, 20, 51–63 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |