Identification, isolation and characterization of a new impurity in cefoxitin drug substance resulting from stress stability studies
Vundavilli Jagadeesh
Kumar
*a,
P.
Badarinadh Gupta
a,
K.
Mukkanti
b,
B.
Shankar Reddy
a,
K. S. R.
Pavan Kumar
*a,
G. K. A. S. S.
Narayan
a,
Yallappa Somappa
Somannavar
a,
G. S.
Siva Kumar
a and
Hemant Kumar
Sharma
a
aAurobindo Pharma Ltd Research Centre, 313, Bachupally, Qutubullapur Mandal, Hyderabad, 500090, Andhra Pradesh, India. E-mail: jagadishkumar28@rediffmail.com; pavan_kothapalli@rediffmail.com
bJ.N.T.University, Kukatpally, Hyderabad, 500085, Andhra Pradesh, India
First published on 16th November 2010
Abstract
A new unknown impurity of cefoxitin formed during a gradient reverse phase high performance liquid chromatography (HPLC) analysis of stress stability samples of the drug substance cefoxitin, and the level of this impurity was found at up to 0.9%. This impurity was identified by LC-MS and characterized by (1H NMR, 13C NMR, LC/MS/MS, elemental analysis and FT-IR). Based on the spectral data, the impurity was named as, 3-[[(2R,3S)-[3-methoxy-3-N-[2-(thiophen-2-yl)acetamido]]-4-oxoazetidin-2-ylthio]-2-[(carbamoyloxy)methyl]]-acrylic acid. The structure of this impurity was also established unambiguously, prepared by isolation and co-injected into HPLC to confirm the retention time. To the best of our knowledge, this impurity has not been reported elsewhere. Structural elucidation of the impurity by spectral data is discussed in detail.
1 Introduction
Cefoxitin is classified as a semisynthetic second generation cephalosporin1 which like other cephalosporins and penicillins, kills bacteria by interfering in the synthesis of the bacteria cell wall. It is derived by chemical modification of the naturally-occurring cephamycin, which gives cefoxitin its high degree of resistance to inactivation by bacterial cephalosporinases. Cefoxitin has been proven to be effective against such bacteria as: Serratis marcescens, Bacteroides fragilis and Proteus morganii.2,3 Cefoxitin is active in vitro against both gram positive and gram negative organisms4 and is therapeutically important because of its resistance to destruction by bacterial β-lactamase.5–7 From pharmacokinetics, it is distributed rapidly between serum and tissues.8 At room temperature cefoxitin is stable for at least three years when protected from moisture. At elevated temperature, the solid exhibits a biphasic decomposition pattern typified by an initial more rapid decomposition period followed by a slower decay period.9 Chemically cefoxitin is (6R,7S)-3-[(carbamoyloxy)methyl]-7-methoxy-8-oxo-7-[[(thiophen-2-yl)acetyl]amino]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate. The empirical formula is C16H17N3O7S2 and molecular weight is 427.4. In this study, cefoxitin drug substance was subjected to stress stability conditions as per International Conference on Harmonization (ICH)10 and the samples were analyzed by HPLC, one unknown impurity was detected along with known pharmacopoeia impurities.11 An impurity profile study has to be carried out for any final product as per the regulatory requirements to identify and to characterize all the unknown impurities.12 To date no mention is available regarding this impurity in the literature to the best of our knowledge. The present work deals with identification, isolation and structural elucidation of this impurity.2 Experimental
2.1 Chemicals, reagents and samples
The investigated samples of cefoxitin and crude samples were procured from APL Research Centre laboratories (a division of Aurobindo Pharma Ltd., Hyderabad.). Analytical reagent (AR grade) sodium dihydrogen orthophosphate dihydrate, dipotassium hydrogen orthophosphate, ammonium acetate, orthophosphoric acid and trifluoroacetic acid; HPLC grade acetonitrile and methanol; IR spectroscopy grade potassium bromide were procured from Merck (India) Limited, AR grade formaldehyde was procured from Sigma-Aldrich (India) and pure milli-Q water was used with a millipore purification system.2.2 High performance liquid chromatography
A Waters Alliance 2695 separation module equipped with 2996 photodiode array detector with Empower pro data handling system [Waters corporation, Milford, MA 01757, USA] was used. The analysis was carried out on a stainless steel column 250 mm long, 4.6 mm internal diameter filled with phenyl groups chemically bonded to porous silica particles of 5 μm diameter [Zorbax SB-Phenyl column (make: Agilent.)] maintained at temperature 30 °C. Mobile phase A was a phosphate buffer, prepared by dissolving 4.0 g of sodium dihydrogen orthophosphate dihydrate in 1000 ml water, adjusted to pH 3.2 with orthophosphoric acid. Mobile phase B was acetonitrile and methanol mixed in the ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v. Diluent was prepared by dissolving 0.7 g of dipotassium hydrogen orthophosphate in 1000 ml water, adjusted to pH 6.8 with diluted orthophosphoric acid solution. Flow rate was kept as 1.0 ml min−1, injection volume was 20 μl, chromatographic data acquisition time for 65 min and UV detection was carried out at 235 nm. The pump was in gradient mode and the program was as follows: Time (min)/A (v/v): B(v/v); T0.01/90:10, T35/80:20, T60/55:45,T65/20:80,T66/90:10, T75/90:10.
:50 v/v. Diluent was prepared by dissolving 0.7 g of dipotassium hydrogen orthophosphate in 1000 ml water, adjusted to pH 6.8 with diluted orthophosphoric acid solution. Flow rate was kept as 1.0 ml min−1, injection volume was 20 μl, chromatographic data acquisition time for 65 min and UV detection was carried out at 235 nm. The pump was in gradient mode and the program was as follows: Time (min)/A (v/v): B(v/v); T0.01/90:10, T35/80:20, T60/55:45,T65/20:80,T66/90:10, T75/90:10.
2.3 Preparative liquid chromatography
A Shimadzu LC-8A preparative liquid chromatograph equipped with SPD-10A VP, UV-Vis detector [Shimadzu corporation, Analytical Instruments Division, Kyoto, Japan] was used. Hyperprep HS C18 500 mm long, 30 mm i.d., make: Thermo Scientific preparative column packed with 10 μm particle size was employed for isolation of impurity. The mobile phase A consisted of 0.2% trifluoroacetic acid solution in water and mobile phase B was acetonitrile. Flow rate was set as 30 ml min−1 and UV detection was carried out at 235 nm.The gradient program was as follows, Time (min)/A(v/v):B(v/v); T0.01/100:0, T30/95:05, T45/90:10,T55/85:15,T65/80:20,T75/20:80.
2.4 LC/MS/MS
LC/MS/MS analysis was carried out using a Perkin Elmer triple quadrupole mass spectrometer (API 2000, PE SCIEX) coupled with a Shimadzu HPLC equipped with SPD 10 AT VP UV-Vis detector and LC 10 AT VP pumps. Analyst software was used for data acquisition and data processing. The turbo ion spray voltage was maintained at 5.5 kv and temperature was set at 375 °C. The auxiliary gas and curtain gas used was high purity nitrogen. Zero air was used as the nebulizer gas. LC-MS spectra were acquired from m/z 100–1000 in 0.1 amu steps with 2.0 s dwell time. The analysis was carried out using Zorbax SB Phenyl, 250 mm long, 4.6 mm i.d., 5μm particle diameter column. Mobile phase A consisted of 0.1% trifluoroacetic acid solution in water and acetonitrile and methanol were mixed in the ratio of 50:50 v/v and used as mobile phase B. Flow rate was kept as 1.0 ml min−1, UV detection was carried out at 235 nm and data acquisition time was 60 min. The gradient program was as follows, Time (min)/A(v/v):B(v/v); T0.01/80:20, T25/70:30, T35/60:40,T45/20:80, T55/10:90, T57/80:20, T65/80:20.
2.5 NMR
1H NMR and 13C NMR experiments were performed on Bruker 300 MHz NMR spectrometer [Bruker AG Industries, Faellanden, Switzerland] using deuterated dimethylsulfoxide (DMSO-d6) as solvent and tetramethylsilane (TMS) as an internal standard.2.6 FT-IR
FT-IR spectra were recorded as KBr pellet on a Perkin-Elmer instrument model-spectrum one.2.7 Stability studies
As per the International Conference on Harmonization, stress testing is to be carried out to identify the likely degradation products or to elucidate the inherent stability characteristics of the active substance.10 In this study, cefoxitin was subjected to following stress conditions.Thermal stress: sample was subjected to dry heat at 105 °C for 120 h.
Hydrolytic conditions:
(a) Acidic hydrolysis: sample solution was mixed with 0.2M hydrochloric acid solution and exposed at 85 °C for 15 min.
(b) Alkaline hydrolysis: sample solution was mixed with 0.05M sodium hydroxide solution and exposed at 85 °C for 10 min.
Oxidative condition: sample solution was mixed with 30% H2O2 solution for 20 min at room temperature.
Photolytic condition: the sample was exposed to photolytic degradation i.e., 10 K Lux for 120 h.
Humidity condition: the sample was exposed to degrade under 90% RH at 25 °C for 120 h.
3 Results and discussion
3.1 Detection and identification of impurity
The sample solutions described under stress stability conditions were diluted to the required concentration (2 mg ml−1) and injected into HPLC using analytical conditions. In oxidative photolytic and humidity degradation chromatograms, one unknown peak was identified at a relative retention time of about 0.94 min with respect to the cefoxitin peak along with pharmacopoeial impurities The same samples were subjected to LC-MS analysis using conditions as described in the LC/MS/MS section to identify the mass of the impurity. The mass of the impurity was 415.8 recorded in the positive ionization mode. This impurity was isolated by preparative HPLC and co-injected with cefoxitin samples into HPLC to confirm the relative retention time. The typical representative HPLC chromatograms of cefoxitin spiked with pharmacopoeial impurities including the new impurity and cefoxitin spiked with only the new impurity are shown in Fig.1a and 1b, respectively.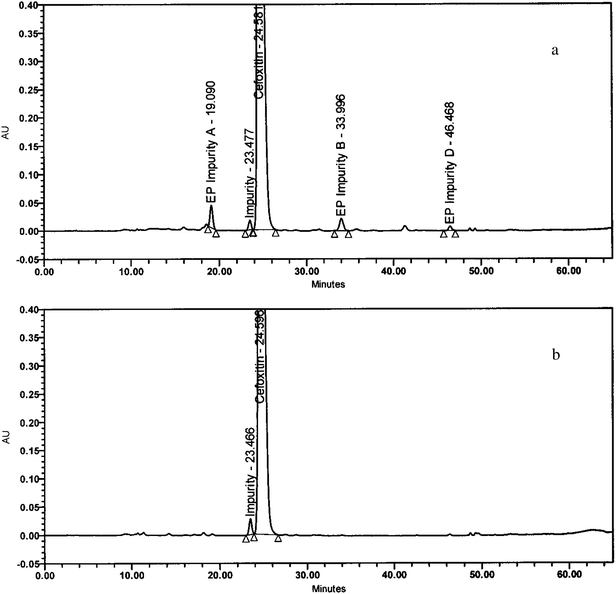 | ||
| Fig. 1 (a) A typical representative HPLC chromatogram of the drug substance cefoxitin spiked with pharmacopoeial impurities including the new impurity. (b) A typical representative HPLC chromatogram of cefoxitin drug substance spiked with the new impurity. | ||
3.2 Isolation of impurity by preparative HPLC
Cefoxitin sample was exposed to degradation under 90% RH at 25 °C for 120 h and analyzed by HPLC, the required impurity was observed at around 0.9%. This enriched sample (about 1.0 g) was taken in a beaker and about 10 ml of 10% w/v ammonium acetate aqueous solution was added to dissolve it. This solution was loaded into the preparative column using the conditions mentioned in the preparative liquid chromatography section. Fractions of impurity ≥ 95% were pooled together, and concentrated on a rotavapour to remove solvent (organic). Concentrated fractions were loaded into a preparative column and the eluent was treated with water to remove buffer used in isolation. Finally, the column was washed with water and acetonitrile in the ratio of 20:80 v/v. Again eluent was concentrated using rotavapour to remove acetonitrile. The aqueous solution was lyophilized using freeze dryer (Virtis Advantage 2XL). The impurity obtained was an off-white powder in description and the chromatographic purity was about 95.0% determined by the HPLC method.
3.3 Structural elucidation of impurity
ESI mass spectrum of the impurity in positive ion mode displayed a molecular ion peak at m/z 415.8 amu [(MH)+], which is 12 amu less than that of the cefoxitin protonated molecular ion. Based on elemental analysis, theoretical values C, 43.41; H, 4.13; N, 10.13; S, 15.45 Found: C, 43.37; H, 4.09; N, 10.11 and S, 15.41 suggests that the elemental composition of this impurity was C15H17N3O7S2 and molecular weight as 415. In 1H NMR and 13C NMR spectra of the impurity chemical shift (δ) values of impurity were similar to values of cefoxitin except at cephem moiety. Moreover, when close inspection of structures of cefoxitin and impurity (Fig. 2a and 2b) with 1H NMR and 13C NMR data (see Table 1), an additional signal was observed at δ 7.17 and δ 136.2 corresponding to acrylic –CH at position 5 instead of AB quartet signal at (δ 3.03, δ 3.40) and δ 25.7 corresponds to –CH2 signal. Absence of a 7th position signal in 13C NMR spectrum of impurity reveals the quaternary carbon deficiency; and additionally in 1H NMR spectrum at 19th position, chemical shift δ 7.76 was observed and corresponds to –NH signal which was exchangeable, when compared with cefoxitin. In 1H NMR spectra, additionally substantial downfield chemical shift was observed at 6th position from δ 4.93 to δ 5.20 and 13C NMR spectrum shows an up field shift was observed from δ 62.8 to δ 58.9. In 13C NMR spectrum, a comparative down field shifts were observed at positions 8 and 9 from δ 162.5, δ 160.3 to δ 168.4, δ 164.1 respectively and also up field shifts were observed at 10th position from δ 95.1 to δ 88.6. From the above interpretation, we suspected intramolecular addition and elimination could be occurring in the cephem moiety, discussed in section 3.4. The chemical structures of cefoxitin and impurity are given in Fig.2a and 2b and 1H, 13C NMR chemical shift values and DEPT assignments are presented in Table 1. Subsequently, from the mass spectrum, mass fragments i.e. m/z 355, 290, 276, 211 and 140 supported the structure of the impurity and a probable fragmentation pattern of the impurity are drawn in Fig.3. In addition, this impurity was confirmed by FT-IR spectral data. A comparative FT-IR spectral data of the impurity with cefoxitin was presented in Table 2. Based on the above spectral data, the impurity was chemically proposed as 3-[[(2R,3S)-[3-methoxy-3-N-[2-(thiophen-2-yl)acetamido]]-4-oxoazetidin-2-ylthio]-2-[(carbamoyloxy)methyl]]-acrylic acid.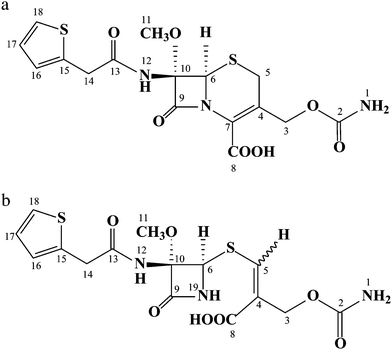 | ||
| Fig. 2 (a) Chemical structure of cefoxitin and (b) chemical structure of the new impurity. | ||
| Positiona | Cefoxitin | Impurity | ||||
|---|---|---|---|---|---|---|
| 1H δ (ppm), multiplicity | 13C δ (ppm) | DEPT | 1H δ (ppm), multiplicity | 13C δ (ppm) | DEPT | |
| a s, singlet; d, doublet; dd, doublet of doublet; m, multiplet; brs, broad singlet; q,quartet; t, triplet, ABq, AB quartet. b a, refer chemical structures (Fig. 2a and 2b) for numbering. | ||||||
| 1 | 6.56 (brs, 2H) | — | — | 6.48 (brs, 2H) | — | — |
| 2 | — | 156.3 | C | — | 157.3 | C |
| 3 | 4.66 & 4.72 (ABq, 2H) | 61.9 | CH2 | 4.47 & 4.58 (ABq, 2H) | 62.0 | CH2 |
| 4 | — | 125.5 | C | — | 125.6 | C |
| 5 | 3.03 & 3.40 (ABq, 2H) | 25.7 | CH2 | 7.17 (s, 1H) | 136.2 | CH |
| 6 | 4.93 (s, 1H) | 62.8 | CH | 5.20 (d, 1H) (J = 4.12 Hz) | 58.9 | CH |
| 7 | — | 125.8 | C | — | — | — |
| 8 | — | 162.5 | C | — | 168.4 | C |
| 9 | — | 160.3 | C | — | 164.1 | C |
| 10 | — | 95.1 | C | — | 88.6 | C |
| 11 | 3.34 (s, 3H) | 52.5 | CH3 | 3.20 (s, 3H) | 54.1 | CH3 |
| 12 | 9.36 (s, 1H) | — | — | 8.51 (s, 1H) | — | — |
| 13 | — | 170.4 | C | — | 170.5 | C |
| 14 | 3.76 & 3.84 (ABq, 2H) | 35.9 | CH2 | 3.79 & 3.87 (ABq, 2H) | 36.9 | CH2 |
| 15 | — | 136.5 | C | — | 137.2 | C |
| 16 & 17 | 6.94–6.96 (m, 2H) | 126.5 & 126.4 | 2 × CH | 6.95–6.98 (m, 2H) | 125.2 & 126.9 | 2 × CH |
| 18 | 7.37 (dd, 1H) | 124.9 | CH | 7.38 (dd, 1H) (J = 4.4 & 1.65 Hz) | 125.7 | CH |
| 19 | — | — | — | 7.76 (d, 1H) (J = 5.2 Hz) | — | — |
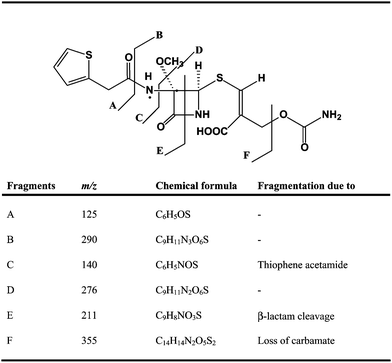 | ||
| Fig. 3 Fragmentation pattern of the impurity. | ||
| IR (KBr) absorption bands (vmax/cm−1) | |||
|---|---|---|---|
| Cefoxitin | Impurity | ||
| a s: strong; m: medium; br: broad. | |||
| 3241–3425 (m) | various NH stretch | 3200–3459 (m) | various NH stretch |
| 2955 (br) | –OH stretch | 3031(br) | –OH stretch |
| 1792 (s) | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (β lactam) stretch O (β lactam) stretch |
1780 (s) | CO (β lactam) stretch |
| 1717 (s) | CO (carboxyl) stretch |
1714 (s) | CO (carboxyl) stretch |
| 1686 (s) | CO (carbamate) stretch |
1660 (s) | CO (carbamate) stretch |
| 1666 (s) | CO (amide) stretch |
1651 (s) | CO (amide) stretch |
| 1426 (m) | various –CH2 blends | 1439 (m) | various –CH2 blends |
| 1084 (s) | carbamate CH2 blends | 1057 (s) | carbamate CH2 blends |
| 1560 (m) | CC stretch |
||
Conclusion
A new unknown impurity was formed in cefoxitin drug substance resulting from stress stability studies and identified by HPLC and LC-MS. The impurity was isolated, characterized by various spectroscopic techniques, NMR, LC/MS/MS and FT-IR and the probable structure of impurity is proposed.Acknowledgements
The authors gratefully acknowledge the management of APL Research Centre (a division of Aurobindo Pharma Ltd), for allowing us to carry out the present work. The authors are also thankful to the colleagues of Analytical Research Department and Chemical Research Department for their co-operation.References
- M. B. Regazzi, G. Chirico, D. Cristiani, G. Rondini and R. Rondanelli, Eur. J. Clin. Pharmacol., 1983, 25, 507–509 CrossRef CAS.
- C. P. Page, J. M. Bohnen, J. R. Fletcher, A. T. McManus, J. S. Solomkin and D. H. Wittmann, Arch. Surg., 1993, 128(1), 79–88 Search PubMed.
- L. A. Wheeler, M. de Meo, B. D. Kirby, R. S. Jerauld and S. M. Finegold, J. Chromatogr., 1980, 183(3), 357–362 CrossRef CAS.
- H. G. Adams, G. A. Stilwell and M. Turck, Antimicrob. Agents Chemother., 1976, 9(6), 1019–1024 CAS.
- G. Darland and J. Birnbaum, Antimicrob. Agents Chemother., 1977, 11(1), 725–734 CAS.
- H. R. Onishi, D. R. Daoust, S. B. Zimmerman, D. Hendlin and E. O. Stapley, Antimicrob. Agents Chemother., 1974, 5(1), 38–48 CAS.
- H. C. Neu, Antimicrob. Agents Chemother., 1974, 6(2), 170–176 CAS.
- R. P. Buhs, T. E. Maxim, N. Allen, T. A. Jacob and F. J. Wolf, J. Chromatogr., A., 1974, 99, 609–618 CrossRef CAS.
- E. R. Oberholtzer and G. S. Brenner, J. Pharm. Sci., 1979, 68(7), 863–866 CrossRef CAS.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonized Tripartite Guideline. Stability Testing of New Drug Substances and Products Q1A(R2), Step 4 2003.
- European Pharmacopoeia, 6th edn, European Directorate for the Quality of Medicines Strasbourg, France, 2008, 1455–1457 Search PubMed.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonized Tripartite Guideline. Impurities in New Drug Substances Q3A(R2), Step 4 2006.
| This journal is © The Royal Society of Chemistry 2011 |