Disposable manganese oxide screen printed electrodes for electroanalytical sensing
Biljana R.
Šljukić
*a,
Rashid O.
Kadara
b and
Craig E.
Banks
*b
aFaculty of Physical Chemistry, University of Belgrade, Studentski trg 12, 11158, Belgrade, Serbia. E-mail: biljka@ffh.bg.ac.rs
bFaculty of Science and Engineering, School of Biology, Chemistry and Health Science, Division of Chemistry and Materials, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, Lancashire, UK. E-mail: c.banks@mmu.ac.uk
First published on 22nd November 2010
Abstract
Manganese dioxide screen printed graphite electrodes for electro-analytical sensing purposes have been fabricated. The prepared sensors exhibit attractive performances as electro-catalysts for the sensing of nitrite ions and ascorbic acid with detection limits comparable or lower than those obtainable with other electrochemical sensors. The manganese dioxide platforms are also explored towards the electrochemical reduction of oxygen which has importance in electrochemical energy storage. While the platforms are found to operate by a four-electron reduction producing water (rather than hydrogen peroxide which is undesirable), a large overpotential, compared to other possible electrode compositions reported in the literature, is required. Nevertheless, the promising results towards the electro-analytical sensing of nitrite ions and ascorbic acid and also given the disposable nature and scale of economies make these electrodes useful as sensor platforms.
Introduction
Heterogeneous carbon electrodes consisting of non-conducting liquid or solid matrix, into which conductive carbon particles are implanted, such as carbon paste electrodes (CPE), solid carbon composite electrodes (CCE), and screen printed carbon electrodes (SPCE) are frequently employed as electro-analytical sensors.1,2 It is their relatively low cost, chemical inertness, wide working potential window, rich surface chemistry of the carbon employed allowing chemical derivatization and modification, that make these electrodes suitability for various applications.3,4However, these electrodes exhibit slow electron transfer towards target analyte which is commonly improved by their modification. So far, noble metals have shown the best overall catalytic performance and played a dominating role as electro-catalysts. Despite their advantages of high catalytic activity, electronic conductivity and stability, their high cost is a serious difficulty for commercial applications. Numerous less expensive materials are successfully used to replace expensive metal materials. Among the more promising oxide catalysts are the manganese oxides (MnOx) due to their prominent advantages of abundance, low cost, environmental friendliness, and considerable electro-catalytic activity. Manganese dioxide in particular can be produced efficiently and relatively inexpensively on a commercial scale by electro-deposition of manganese dioxide onto a titanium anode from a hot aqueous solution of sulfuric acid and manganese sulfate.5 The electrochemical properties of manganese dioxide have been summarized in the literature6 and it is believed that the presence of H atoms and OH groups is responsible for their electrochemical activity. Moreover, their composition almost never corresponds strictly to MnO2 but is somewhat substoichiometric and several modifications exist. Normally, manganese dioxide consists of manganese in lower valence states such as Mn(II,III), together with the dominant Mn(IV) state. Although having excellent activity, the unloaded manganese oxides suffer from very low surface areas. Loading manganese oxides onto a suitable support, either by direct synthesis or post-grafting, provides highly dispersed surface oxides. In our previous work MnO2 has been used as an electro-catalyst for the electro-analytical sensing of nitrite ions, ascorbic acid and hydrogen peroxide7,8 where carbon powder has been chemically modified by manganese dioxidevia a wet impregnation method, including high or low temperature procedure.
Recently we have demonstrated that metal oxide screen printed electrodes can be readily fabricated which offer greater simplification over currently adopted fabrication methodologies and also have the advantage of scales of economy, portability and similar sensitivities and linear ranges comparable to nanoparticle modified counterparts.9–11 Following on with this strategy, we report the first manganese dioxide modified screen printed carbon platforms and evaluate the sensors electro-analytical performance towards the sensing of nitrite ions and ascorbic acid as well as for electro-catalysis of the oxygen reduction reaction in alkaline solutions. Note that we find examples of manganese dioxide screen printed electrodes used in energy storage applications but very few electro-analytical applications;12,13 the low cost through scales of economy and disposable nature of these electrodes make them extremely attractive as sensors. In the electro-analytical context, the forerunner of screen printed electrodes were carbon paste electrodes. Vytřas and co-workers modified carbon pastes with manganese dioxide films for hydrogen peroxide sensing.14–16 For application into flow injection analysis, these sensors were transferred to screen printed configurations and also modified with glucose oxidase to facilitate biosensors.
In this paper we explore the electrochemical performance of our fabricated MnO2 screen printed electrode with the example of sensing of nitrite and ascorbic acid due to their importance in food and pharmaceutical industry and environment. It is almost impossible to determine these analytes by direct electrochemical oxidation on many conventional electrodes as undesirably large overpotentials are usually required.17–19 In order to improve their determination, the operating potentials should be efficiently lowered which can be achieved by modification of the electrode surfaces with suitable electro-catalysts. Oxygen reduction reaction, where molecular oxygen is electro-catalytically reduced, is vital in the fields of electrochemical energy storage and conversion including fuel cells and metal–air batteries.20–22 The oxygen reduction reaction proceeds either as a direct four-electron process or a two-electron process. Production of water by a four-electron oxygen reduction is used both in fuel cells and gas sensors, while hydrogen peroxide is produced by a two-electron reduction of oxygen which is undesirable.
Experimental
All chemicals and reagents used in this work were of the analytical grade and used as received without further purification. All aqueous solutions were prepared with distilled water. pH 7 phosphate buffer was used as supporting electrolyte for sensing of ascorbic acid, while 1 mM HClO4 + 1M NaClO4 was used as supporting electrolyte for determination of nitrite ions. The supporting electrolyte for study of oxygen reduction reaction was 0.1 M aqueous KOH solution which was deaerated by bubbling in nitrogen gas (Messer). Alternatively, oxygen gas (Messer, purity 99.995%) was bubbled in for 10 min before each investigation. In addition, oxygen-saturated atmosphere was maintained throughout the electrochemical experiments. All electrochemical measurements were conducted at Gamry instrument using a 10 cm3cell at room temperature.Screen-printed carbon electrodes were fabricated in-house with appropriate stencil designs using a microDEK 1760RS screen-printing machine (DEK, Weymouth, UK). A carbon–graphite ink (Gwent Materials, UK) was first screen printed onto a polyester flexible film (Autostat, 250 µm thickness). This layer was cured in a fan oven at 60 °C for 30 minutes. Next a silver/silver chloride reference electrode was included by screen printing Ag/AgCl paste (Gwent Electronic Materials Ltd, UK) onto the plastic substrate. Last a dielectric paste ink (Gwent Electronic Materials Ltd, UK) was printed to cover the connection and define the 3 mm diameter graphite working electrode. After curing at 60 °C for 30 minutes the screen printed electrode is ready to use. Manganese oxide screen printed electrodes were fabricated as described above but with commercially purchased manganese oxide, that was ball-milled to a fine consistency, and efficiently mixed into the ink formulation prior to screen printing. Note that in doing so, the rheology of the ink changes and the addition of solvents is needed along with modification of the standard printing parameters. Increasing amounts of manganese oxide were incorporated into the screen printed electrodes over the range of 0–10% (MP/MI), where MP is the mass of particulate and MI is the mass of ink formulation used in the printing process. Scanning electron microscope (SEM) images and surface element analysis were obtained with a JEOL JSM-5600LV model having an energy-dispersive X-ray microanalysis package.
Results and discussion
Sensing of nitrite
We first consider the electro-analytical sensing of nitrite ions in aqueous solution using the manganese oxide screen printed carbon (MnO2-SPC) electrode sensing platforms using linear sweep voltammetry. The response of a 2% (MP/MI) MnO2-SPC electrode was recorded in a solution of 1 mM HClO4 + 1 M NaClO4 over the potential range of 0.0 V to +1.25 V at a scan rate of 50 mV s−1. As shown in Fig. 1, no voltammetric peak was observed in the absence of nitrite ions but following the addition of 1 mM nitrites, a large and easily distinct voltammetric peak corresponding to the electrochemical oxidation of nitrite ions could be observed at ca. +0.63 V. Note that at bare carbon electrode surfaces the oxidation of nitrite occurs at higher positive potentials.23 Moreover, in comparison to literature reports, using linear sweep voltammogram at MnO2 carbon epoxy composite electrodes under the same conditions exhibited a voltammetric peak at ca. 0.3 V more positive potential7,8 compared to that observed for oxidation of nitrite ions (viz.Fig. 1) at the MnO2-SPC electrodes. The response of differing % (MP/MI) MnO2-SPC electrodes towards the electro-analytical sensing of nitrite was explored where it was found that as the % (MP/MI) manganese dioxide content in the screen printed sensor was increased, the background current also enlarged until at the 10% (MP/MI) MnO2-SPC electrodes, the large background overcomes the electro-analytical signal. This has been observed many times before in our prior work which is likely due to the reduced number of conductive pathways throughout the electrode.9,10SEM was used to explore the 2% (MP/MI) MnO2-SPC electrode where a relatively rough surface was observed but is not distinctly different to that expected for a non-modified screen printed electrode9,10 and did not provide any useful information. However, we know that the micron sized manganese oxide likely resides in the carbon layer as random dispersed micron sized particles, or larger, which are clearly exposed to the electrolyte solution permitting the electrochemical signal towards nitrite as observed in Fig. 1. Using a fixed concentration of nitrite (500 µM) as a voltammetric probe, the % RSD (n = 20) was found to correspond to 7.2%.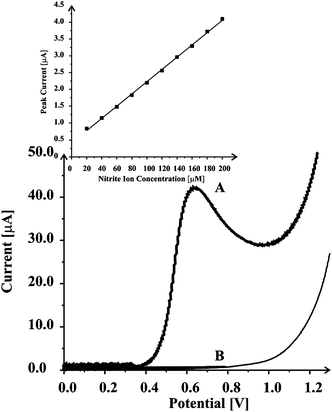 | ||
| Fig. 1 Comparison of linear sweep voltammograms of MnO2-SPC sensing platform in 1 mM HClO4 + 1 M NaClO4 (B) and in 1 mM NaNO2 solution (A) at a scan rate of 50 mV s−1. The standard addition plot obtained by spiking the solution with 20 µM nitrite solution is also shown (inset). | ||
Next, 20 µM additions of nitrite were made into a HClO4 + 1 M NaClO4 and the inset of Fig. 1 depicts the standard addition line; the limit of detection for nitrite was calculated via 3σ24 using data obtained from this plot and found to correspond to 2.5 µM. Note that this detection limit is comparable or lower than detection limits obtained with existing methodologies, both electrochemical methods25–31 and other techniques.32–34 This indicates that MnO2-SPC sensing platforms can potentially be used for monitoring of the concentration of nitrite in drinking water where the maximum allowed nitrite level is 2.7 µM.35 For application of the platforms for the detection of nitrite ions in real samples it is necessary to recognize the possible interference of species being present in complex media, particularly ascorbic acid and ions like nitrates.
Ascorbic acid sensing
We now turn to exploring the MnO2-SPC electrode towards the detection of ascorbic acid. Cyclic voltammograms of 2% (MP/MI) MnO2-SPC electrode were initially run in 1 mM ascorbic acid aqueous solution, with a distinct peak observed at ca. +0.24 V (Fig. 2). No peak was observed in the absence of ascorbic acid. Note that the ascorbic acid oxidation peak is observed at the potentials somewhat lower than the ones obtain with non-modified carbon electrodes.36 Furthermore, note that the peak of ascorbic acid oxidation appears at the potential that is ca. 0.4 V less positive than the obtained nitrite ion oxidation peak. This demonstrates that manganese dioxide screen printed carbon platforms can be used for sensing of both nitrite ions and ascorbic acid even if they are simultaneously present in a sample. Again, the % (MP/MI) MnO2-SPC electrode was explored and again it was found that 2% (MP/MI) MnO2-SPC electrode was the optimum. Subsequently, the blank solution was spiked with 20 µM additions of ascorbic acid and the observed voltammetric peak current was found to be linearly proportional to the concentration of ascorbic acid in the solution and limit of detection was evaluated using 3σ method. Detection limit for ascorbic acid using MnO2-SPC platform was found to be 2.8 µM. The data from the repeated experiments revealed good reproducibility with 4.8% deviation. This detection limit of 2.8 µM for ascorbic acid obtained with the MnO2-SPC electrode is comparable to the ones obtained with the MnO2 carbon epoxy composite electrodes7,8 and lower or comparable with the LODs obtained with the existing methodologies36–39 that range from 0.08 to 71 µM.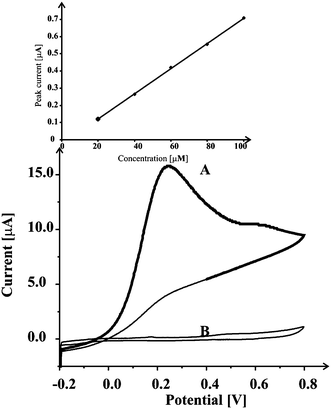 | ||
| Fig. 2 Cyclic voltammograms of MnO2-SPC sensing platform in pH 7 phosphate buffer (B) and in 1 mM ascorbic acid solution (A) at a scan rate of 50 mV s−1. Response for 20 µM ascorbic acid additions to a blank solution is also included. | ||
Further investigation has been performed in order to confirm possible application of MnO2-SPC electrodes for sensing of nitrite ions and ascorbic acid when both analytes are present in a sample. Linear sweep voltammogram was recorded in solution containing milli-molar concentrations of ascorbic acid and nitrite ions. The solution was made with tap water in order to consider utility of the platforms in real samples. Without any optimization of conditions (for example, pH optimization), two distinct peaks could be observed at the recorded voltammogram, Fig. 3. The first peak at ca. +0.13 V corresponds to the oxidation of ascorbic acid and the second peak, at ca. +0.75 V corresponds to the oxidation of nitrite ions. The obtained results showed that MnO2-SPC platforms can be used for sensing of nitrite ions and ascorbic acid when present in high concentrations in the same sample.
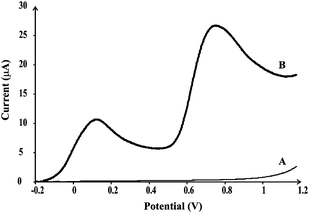 | ||
| Fig. 3 Linear sweep voltammograms of MnO2-SPC sensing platform in tap water without (A) and when ascorbic acid and nitrite ions are present (B) recorded at a scan rate of 50 mV s−1. | ||
Oxygen reduction reaction
The catalytic activity of the manganese dioxide screen printed carbon systems for the oxygen reduction reaction was first characterized by using the catalytic electrode as the working electrode in nitrogen- and oxygen-saturated 0.1 M KOH solutions, respectively. Fig. 4 shows cyclic voltammograms in the two solutions at a scan rate of 50 mV s−1. Significant reduction current is observed, with a reduction peak at ca.−0.82 V in the oxygen-saturated electrolyte. In the nitrogen-saturated electrolyte, no peak has been observed. From the prominent difference between reduction currents in the two solutions, the significant reduction current in the oxygen-saturated solution must be due to catalyzed oxygen reduction reaction. For comparison, a small reduction current is observed in the solution saturated with ambient air due to the electrochemical reduction of dissolved oxygen. Cyclic voltammograms of the manganese dioxide screen printed carbon electrode in oxygen-saturated 0.1 M KOH were recorder at different scan rates in the range from 5 to 100 mV s−1 (Fig. 5). The peak current was found to linearly increase with square root of scan rate employed, indicating that the oxygen reduction at these electrodes is a diffusion-controlled process. Note that two peaks could be seen: at ca. −0.63 and ca. −0.82 V at cyclic voltammograms recorded at lower scan rates. The oxygen reduction reaction on manganese dioxide based electrodes in alkaline media has been previously studied40–42 but the exact mechanism of the reaction remains uncertain. It is believed that two- and four-electron mechanisms of the oxygen reduction reaction are competing on these electrode surfaces. In our case it is likely that the two waves observed at low scan rates are due to MnO2 microdomains (at −0.82 V) and that of the underlying carbon surface (−0.63 V). The number of electrons transferred involved in the oxygen reduction was estimated with Randles–Sevčík equation.43 A geometric area of the working electrode (3 mm diameter) was used since due to the scan rates employed in measurement, diffusional overlap of each MnO2 microdomain will occur such that the diffusion profile is linear in nature. The number of electrons transferred was calculated to be 3.7, suggesting that an apparent four-electron reduction of oxygen was almost obtained at the manganese dioxide screen printed carbon electrodes where the MnO2 microdomains are the electro-active sites. The manganese dioxide screen printed carbon platforms show good electro-catalytic activity for sensing of nitrite ions and ascorbic acid, but require a high overpotential for the electrochemical reduction of oxygen. Though other approaches to oxygen reduction reaction with lower overpotentials are available in the literature, the one presented herein has the advantage that water is the final product, rather than hydrogen peroxide. Given the low cost of each sensor platform, they might be beneficial in fuel cells/supercapacitors.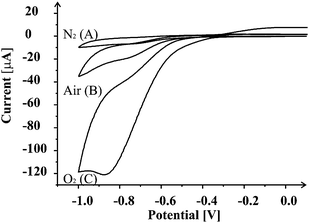 | ||
| Fig. 4 Comparison of cyclic voltammograms of MnO2-SPC platform recorded at a scan rate of 50 mV s−1 in 0.1 M KOH saturated with nitrogen (A), air (B) and oxygen (C). | ||
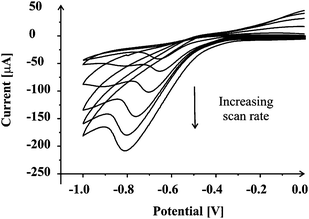 | ||
| Fig. 5 Cyclic voltammograms of MnO2-SPC platform in 0.1 M KOH solution saturated with oxygen recorded at different scan rates. | ||
Conclusions
In conclusion, manganese dioxide screen printed carbon platforms have been fabricated through a facile route. The obtained MnO2-SPC platforms have been examined as catalysts in electro-analysis as well as for electrochemical reduction of oxygen. The sensors required undesirably high overpotential for the oxygen reduction reaction. However, they showed high catalytic activities for electrochemical sensing of nitrite ions and ascorbic acid indicating the potential applications of manganese dioxide screen printed carbon platforms for sensing of the named analytes. Given the low cost and disposable nature, their analytical utility and electro-catalytic effect make them a beneficial choice as a sensor.Acknowledgements
B.Š. would like to thank Ministry of Science and Technological Development of the Republic of Serbia for the support within the project No. 142047.References
- F. Kapteijn, A. D. van Langeveld, J. A. Moulijn, A. Andreini, G. Ramis and G. Busca, J. Catal., 1994, 150, 105 CrossRef CAS.
- T. Grzybek, J. Pasel and H. Papp, Phys. Chem. Chem. Phys., 1999, 1, 341 RSC.
- R. L. McCreey and K. K. Cline, in Laboratory Techniques in Electroanalytical Chemistry, ed. P. T. Kissinger and W. R. Heinemann, Dekker, New York, 2nd edn, 1996, p. 293 Search PubMed.
- J. Wang, Analytical Electrochemistry, VCH, New York, 1994 Search PubMed.
- C. B. Ward, A. I. Walker and A. R. Taylor, Prog. Batteries Battery Mater., 1992, 11, 40 Search PubMed.
- J. P. Brenet, J. Power Sources, 1979, 4, 183 CrossRef CAS.
- B. Šljukić and R. G. Compton, Electroanalysis (N. Y.), 2007, 19, 1275 CrossRef CAS.
- C. E. Langley, B. Šljukić, C. E. Banks and R. G. Compton, Anal. Sci., 2007, 23, 165 CrossRef.
- N. A. Choudhry, D. K. Kampouris, R. O. Kadara, N. Jenkinson and C. E. Banks, Anal. Methods, 2009, 1, 183 RSC.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Electroanalysis, 2009, 21, 2410 CAS.
- P. M. Hallam, D. K. Kampouris, R. O. Kadara, N. Jenkinson and C. E. Banks, Anal. Methods, 2010, 2, 1152 RSC.
- N. W. Beyene, P. Kotzian, K. Schachl, H. Alemuc, E. Turkušić, A. Čopra, H. Moderegger, I. Švancara, K. Vytřas and K. Kalcher, Talanta, 2004, 64, 1151 CrossRef CAS.
- K. Vytřas, I. Švancara and R. Metelka, J. Serb. Chem. Soc., 2009, 74(10), 1021 CrossRef CAS.
- K. Schachl, H. Alemu, K. Kalcher, J. Ježková, I. Švancara and K. Vytřas, Analyst, 1997, 122, 985 RSC.
- K. Schachl, H. Alemu, K. Kalcher, J. Ježková, I. Švancara and K. Vytřas, Anal. Lett., 1997, 30, 2655 CAS.
- K. Schachl, H. Alemu, K. Kalcher, J. Ježková, I. Švancara and K. Vytřas, Sci. Pap. Univ. Pardubice, Ser. A, 1997, 3, 41 Search PubMed.
- A. Y. Chamsi and A. G. Fogg, Analyst, 1988, 113, 1723 RSC.
- J.-B. Raoof, R. Ojani and A. Kiani, J. Electroanal. Chem., 2001, 515, 45 CrossRef CAS.
- J.-M. Zen, D.-M. Tsai and A. S. Kumar, Electroanalysis, 2002, 15, 1171.
- A. J. Appleby and F. R. Foulkes, Fuel Cell Handbook, Van Nostrand Reinhold, New York, 1992 Search PubMed.
- Kinoshita, Electrochemical Oxygen Technology, Wiley, New York, 1992 Search PubMed.
- K. A. S. Arico, S. Srinivasan and V. Antonucci, Fuel Cells (Weinheim, Ger.), 2001, 1, 133 Search PubMed.
- B. R. Kozub, N. V. Rees and R. G. Compton, Sens. Actuators, B, 2010, 143, 539 CrossRef.
- C. M. A. Brett and A. M. O. Brett, Electroanalysis, Oxford Chemistry Primers No. 64, Oxford, 1998 Search PubMed.
- M. J. Moorcroft, J. Davis and R. G. Compton, Talanta, 2001, 54, 785 CrossRef CAS.
- J. Davis, M. J. Moorcroft, S. J. Wilkins, R. G. Compton and M. F. Cardosi, Analyst, 2000, 125, 737 RSC.
- J. Davis and R. G. Compton, Anal. Chim. Acta, 2000, 404, 241 CrossRef CAS.
- M. J. Moorcroft, L. Nei, J. Davis and R. G. Compton, Anal. Lett., 2000, 33, 3127.
- A. Abbaspour and M. A. Mehrgardi, Talanta, 2005, 67, 579 CrossRef CAS.
- J.-L. Chang and J.-M. Zen, Electroanalysis, 2006, 18, 941 CrossRef CAS.
- L. Jiang, R. Wang, X. Li, L. Jiang and G. Lu, Electrochem. Commun., 2005, 7, 597 CrossRef CAS.
- M.-Y. Huang, L. Gan, D.-X. He and Y.-Z. Li, Zhongguo Weisheng Jianyan Zazhi, 2005, 15, 1189 Search PubMed.
- H. Ozmen, F. Polat and A. Cukurovali, Anal. Lett., 2006, 39, 823 CrossRef CAS.
- H. Wu, Zhongguo Weisheng Jianyan Zazhi, 2005, 15, 712 Search PubMed.
- http://www.dwi.gov.uk/regs/infolett/1998/info1298.htm .
- F. Wantz, C. E. Banks and R. G. Compton, Electroanalysis, 2005, 17, 1529 CrossRef CAS.
- X.-L. Luo, J.-J. Xu, W. Zhao and H.-Y. Chen, Anal. Chim. Acta, 2004, 512, 57 CrossRef CAS.
- E. Turkusić, V. Milicević, H. Tahmiscija, M. Vehabović, S. Basić and V. Amidžić, Fresenius' J. Anal. Chem., 2000, 368, 466 CrossRef CAS.
- R. T. Kachoosangi, C. E. Banks and R. G. Compton, Electroanalysis, 2006, 18, 741 CrossRef.
- Y. L. Cao, H. X. Yang, X. P. Ai and L. F. Xiao, J. Electroanal. Chem., 2003, 557, 127 CrossRef CAS.
- F. H. B. Lima, M. L. Calegaro and E. A. Ticianelli, J. Electroanal. Chem., 2006, 590, 152 CrossRef CAS.
- I. Roche and K. Scott, J. Electroanal. Chem., 2010, 638, 280 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons Inc, New York, 2nd edn, 2001 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |