The ways to the trace level analysis in infrared spectroscopy
Ana
Gonzalvez
a,
Salvador
Garrigues
a,
Miguel
de la Guardia
*a and
Sergio
Armenta
b
aAnalytical Chemistry Department, University of Valencia, 50th Dr Moliner Street, 46100, Burjassot, Spain. E-mail: miguel.delaguardia@uv.es; Fax: +34-963544845; Tel: +34-963544838
bDepartment of Chemistry, UniversitatAutonoma de Barcelona, EdificiCn, Bellatera 08192, Spain
First published on 18th October 2010
Abstract
The future of infrared (IR) spectroscopy as an analytical technique is assured due to its versatility and its numerous advantages; such as the possibility to obtain molecular specific information for virtually any sample in any state with no treatment or minimal sample preparation. However, spectroscopists are not satisfied with relegating IR spectroscopy just to major and minor component analysis and have been looking at analysis at the trace level too. This review is the recognition of the brilliant research performed during the past two decades and the advances achieved in this area, which have made possible the analysis of contaminants at parts per billion (ppb) levels by IR in different matrices; such as water and soils.
Sensitivity problem of IR procedures
Infrared (IR) spectroscopy is one of the most important and versatile analytical techniques available for today's analytical chemists. The reasons for its great success are based on the fact that it is a rapid and easy to handle methodology which provides molecular specific information for virtually any sample in any state (gases, liquids and solids) due to the different available sampling techniques and the big number of bands presented by the heteroatomic molecules. Another advantage of IR spectroscopy is the possibility to perform both, qualitative and quantitative analysis, of interrogated samples.The IR electromagnetic range can be divided into three different parts: the near-IR region (NIR), from 12500 cm−1 (800 nm) to 4000 cm−1 (2500 nm), the mid-IR (MIR), from 4000 cm−1 (2.5 μm) to 400 cm−1 (25 μm) and the far-IR region (FIR), from 400 cm−1 (25 μm) to 10 cm−1 (1 mm). FIR is scarcely used in analytical chemistry and the low absorbance of overtones and combinations of NIR spectroscopy usually restricts its application range to concentrated liquid and solid samples. However, nowadays, NIR is increasingly accepted as a valuable tool in process analysis in the food, agriculture, pharmaceutical and polymer industries due to the possibilities offered for in-line rapid multi-component quantification but its application to environmental analysis is restricted due to its limited sensitivity. On the other hand, MIR spectroscopy has been the preferred IR range for analytical purposes in environmental applications and thus, this review deals with spectroscopy based procedures related to MIR radiation.
The corresponding MIR bands of a sample spectrum are related to the rotational and vibrational transitions of the molecular bonds and, therefore, the MIR spectrum provides specific structural information in spite of the absence of chromogenic or fluorogenic groups. However, the main drawback as compared to the ultraviolet-visible range is the small extinction coefficient of the IR absorption bands and the strong absorption of water and other commonly used solvents which reduce drastically the optical path, this resulting in the relatively poor detection limits of IR. The strong absorption of water is a major limiting factor due to the losses of spectral information and in terms of sensitivity a feasible pathlength should not exceed 10–20 μm. The water problem can be avoided by using the attenuated total reflectance (ATR) sampling mode using flow cells with a 10–20 μm pathlength in transmission measurements. So, the improvement of the detection limits in MIR spectroscopy has been on the spectroscopist’s mind for a long time.
The analytical quantitative use of IR spectroscopy started with the introduction of the Fourier transform (FT) principle, which dramatically improved IR possibilities to solve a wide range of analytical problems. The high scanning speed and sensitivity of FTIR spectrometers compared with dispersive ones, combined with extended computer facilities1 and the development of powerful laser sources and sensitive detectors,2 have brought MIR from simple qualitative applications to its current state of versatility and popularity. Other advances achieved in this area have been the use of multipass cells, such as White3 or Herriott4 type, featuring total path lengths of tens of meters in absorption measurements in the gas phase.
Strategies to increase sensitivity
To increase the sensitivity of IR procedures, several approaches have been developed in the literature based on i) the combination of preconcentration techniques as solid phase extraction (SPE) or solid phase microextraction (SPME) with IR measurements (see Table 1) and ii) the use of powerful IR sources and improved detection systems. Fig. 1 shows a scheme of the different strategies used to increase the sensitivity of IR measurements. In addition to those preconcentration strategies, alternative techniques such as surface enhanced infrared absorption (SEIRA) spectroscopy could be used to increase the sensitivity of infrared measurements.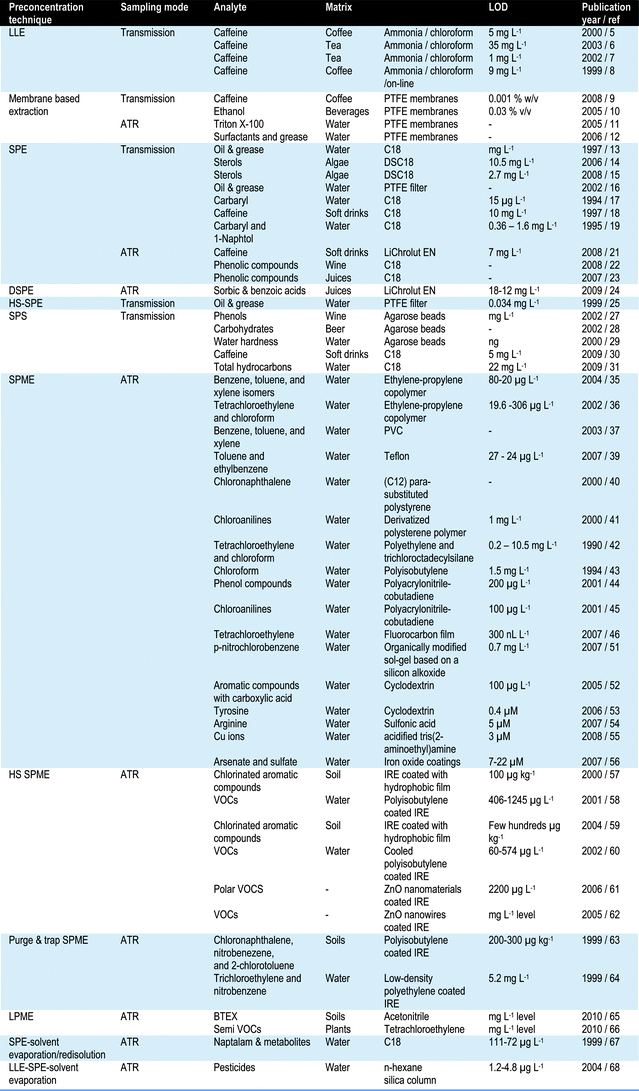
 | ||
| Fig. 1 Strategies employed for improving the IR sensitivity. | ||
Preconcentration strategies
Membrane based extraction has been successfully coupled with ATR spectroscopy for the determination of Triton-X in water samples.11 The developed sequential injection (SI) manifold included a flow cell that allowed the on-line liquid–liquid extraction of the analyte into an organic solvent layer deposited on the ATR surface. The behavior of different organic solvents was evaluated in order to increase the applicability and versatility of the proposed system. The method was successfully applied for the determination of surfactant and oil total indices in industrial degreasing baths12 obtaining a good correlation with a reference methodology based on gravimetric measurements.
An alternative procedure using SPE was developed for the analysis of oil and grease in waters.16 The use of a PTFE filter as a solid phase allows retention of oil and grease and direct analysis of the filter without elution can be performed by transmission measurements with detection limits of 0.034 mg L−1. Although recoveries of diesel oil, pump vacuum oil, olive oil, sunflower oil and mixtures of oils were satisfactory, recoveries of isooctane and benzene gave poor results.
On-line SPE-FTIR procedures involve the use of FIA or SIA manifolds. A FIA manifold was developed in combination with transmission measurements for the determination of carbaryl17 after concentration onto a C18 SPE cartridge and online elution with dichloromethane. The limit of detection of the procedure was 15 ppm (mg L−1) of carbaryl with a relative standard deviation of 8.6%. A similar procedure was developed for the analysis of caffeine in soft drinks eluted with chloroform,18 providing a limit of detection of 10 mg L−1 and a relative standard deviation of 3.5% and an strategy, based on the in-field sampling and on-line elution, provided detection limits of 0.36 mg L−1 for carbaryl and 1.6 mg L−1 for 1-naphthol for the simultaneous determination of these compounds in water samples eluted with chloroform.19 Moreover, an automated method for the determination of organic acids and sugars in soft drinks by using a SI-FTIR manifold including a SPE unit has been successfully developed.20 Organic acids were completely retained on the quaternary amino SPE column whereas sugars passed to the flow cell. The organic acids were subsequently eluted by injection of an alkaline solution.
On the other hand, elution can also be performed with non-chlorinated organic solvents, such as methanol, and even with acidified water, depending on the compounds of interest. In those cases, the preferred sampling method is ATR due the intrinsic characteristics of the technique. For instance, a SPE sorbent material; such LiChrolut EN, located on the internal reflection element (IRE) without using any external coating substance, increased the sensitivity of the system.21 The aforementioned sensor was validated using the determination of caffeine in soft drinks with a sensitivity of 7 mg L−1. Additionally, ATR spectroscopy has been used to differentiate Greek red wines22 and to classify commercial juices23 based on their phenol-rich and sugar-rich extracts after SPE concentration and elution by acidified water and methanol.
A procedure called dispersive SPE has been successfully performed in combination with ATR spectroscopy for the determination of sorbic and benzoic acids in juices.24 The method is based on a dispersive extraction of the analytes from the matrix using a polystyrene-divinylbenzene-based material and a stirring process. The extracted analytes were directly determined in the sorbent with recoveries in the range from 87 to 90%.
The headspace (HS) technique has been also successfully applied in combination with SPE extraction for the analysis of oil and grease in waters.25 Oil and grease can be measured directly in the surface of a PTFE filter with recoveries for various tested oils ranging from 90 to 110%.
When the active solid support, used to pre-concentrate the analyte, is placed inside the measurement cell for the direct measurement of the species of interest, retained onto the solid phase, the technique is called solid phase spectroscopy or solid phase spectrophotometry (SPS).26 This technique has been used to improve detection limits in transmission IR spectroscopy. For instance, flexible agarose beads have been used as the solid phase material in the determination of wine astringency by retaining wine phenols on proteins immobilized in the flow cell,27 carbohydrates in beer employing the beads carrying immobilized maltase28 and water hardness based on an indirect method.29 On the other hand rigid C18 silica beads have been also successfully used to increase the sensitivity of IR methods. The capability of the developed flow through FTIR sensor to determine non-polar molecules, at the parts per million level, was demonstrated on the examples of caffeine,30 with detection limits of 5 mg L−1, and total hydrocarbons in water samples31 with a detection limit of 22 mg L−1.
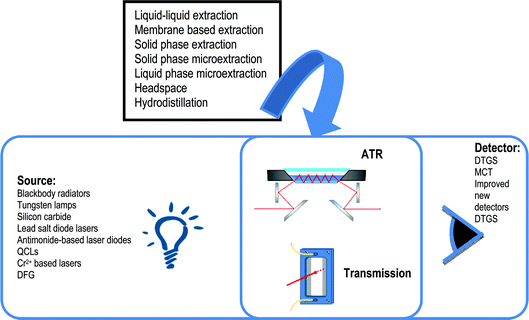
 | ||
| Fig. 2 Solid phase microextraction-ATR-FTIR strategies employed for increasing the limit of detection values of determination through the use of: A) an IRE thin film coating in contact with the sample, B) a HS and C) a purge and trap system for analyte vaporization and enrichment in the coated IR. | ||
Different polymeric materials have been described in the literature for the determination by SPME-FTIR-ATR of contaminants in water at trace levels. For instance, an ATR sensor based on an ethylene-propylene copolymer was developed to determine benzene, toluene, p-xylene, o-xylene, and m-xylene (BTEX) in water samples35 providing detection limits of 45, 80, and 20 μg L−1 for benzene, toluene and all xylene isomers, respectively. The same type of ethylene-propylene copolymer was used for monitoring tetrachloroethylene and chloroform in drinking water providing detection limits of 19.6 and 306 μg L−1, respectively.36 Plasticised polyvinyl chloride (PVC) polymer membranes have been also evaluated for the determination of BTEX compounds in water samples37 observing that the enrichment was much greater in the presence of a plasticizer. In this sense, A.M.S. Silva et al.38 evaluated the effect of different plasticizers on the determination of BTEX in water samples based on transmittance measurements in the MIR region. It was observed that benzene and xylenes do not show interferences, but ethylbenzene and toluene required a multivariate calibration for their determination. Moreover, Teflon coated ATR sensors reported a limit of detection for toluene and ethylbenzene of 27 and 24 μg L−1, respectively.39 Yang and Huang40 compared the performance of various polymers for the detection of chloronaphthalene in aqueous media, showing that alkyl chain (C12) p-substituted polystyrene provides the best sensitivity for PAHs, but with a very slow diffusion. Derivatized polysterene polymers were also used for the determination of chloroanilines in water41 providing detection limits of the order of 1 mg L−1. Different polymers have been tested for tetrachloroethylene and chloroform determination in water42 obtaining detection limits of 0.2 and 2.5 mg L−1 for tetrachloroethylene in low and high density polyethylene, respectively and 10.5 mg L−1 for chloroform in trichloroctadecylsilane coated IRE. On the other hand, using polyisobutylene (PIB) a limit of detection of 1.5 mg L−143 was achieved for chloroform in water samples. Polyacrylonitrile-cobutadiene (PAB) coating has been successfully employed for the determination of phenol compounds44 with detection limits lower than 0.200 mg L−1 and chloroanilines45 with detection limits lower than 0.1 mg L−1. Fluorocarbon films deposited onto IREs have been successfully developed for trace organic contaminant determination in water.46 Tetrachloroethylene was selected as model analyte providing limits of detection below 300 nL L−1.
Environmental applications of ATR sensors (with and without fiber optics) based on IREs coating with polymeric films have been the main area of research and different review papers are available in the scientific literature.47,48 It should be highlighted the sensor called ‘physicochemical mid infrared fiber-optic evanescent-wave (MIR-FEWS)’, which was developed and deeply investigated during the nineties of the last century.49 Analysis of volatile organic pollutants, such as chlorinated or aromatic hydrocarbons, is the area that will probably has the most widespread application of those IR sensor systems, being the development of remote IR fiber-optic sensor for the determination of chlorinated hydrocarbons in water50 a clear example of it.
Moreover, organic-inorganic hybrid materials and cyclodextrins have been developed as IRE coatings for the detection of different polar compounds. Recently, an organically modified sol–gel based on a silicon alkoxide has been used to detect p-nitrochlorobenzene down to 0.7 mg L−151 and cyclodextrin has been also employed for the determination of aromatic compounds with carboxylic acid groups, with detection limits of 0.1 mg L−1.52 Cyclodextrin modified IREs have been successfully used for tyrosine determination in biological fluids53 with a detection limit of 0.4 μM. Arginine was determined using a sulfonic acid modified IRE54 with a detection limit of 5 μM. Moreover, an acidified tris(2-aminoethyl)amine phase was employed for coating the sensing element for the determination of Cu ions in aqueous solutions with a limit of detection of 3 μM.55 Two iron oxide coatings, goethite and an iron sol–gel, were used to determine arsenate and sulfate.56 Goethite provided detection limits of 22 and 7 μM for arsenate and sulfate and the iron sol coating of 2, 6 and 9 μM for arsenate, sulfate, and selenate, respectively.
In spite of the extended use of IRE coated systems, some problems have been enumerated when a polymer-coated IRE was soaked in aqueous solutions for long periods due to: i) changes in the physical properties due to polymer film striping out of the IRE and swelling, ii) diffusion of analytes into the polymer film, that can modify optical properties leading to signals with matrix variances and iii) undissolved particles transferred to the surface of the IREs can provide variations in the analytical signals. To reduce the aforementioned problems in SPME-ATR liquid probes, different methodologies based on HS sampling and purge-and-trap technique have been applied to the ATR sensing method.
Using the HS methodology (see Fig. 2b), chlorinated aromatic and aliphatic compounds have been determined in soil57 and water58 samples using a PIB coated IRE with limits of detection around a few hundreds of ppb. Semivolatile aromatic compounds have been also determined in soil samples59 with detection limits lower than 0.1 mg kg−1. Limits of detection can be also improved by cooling the PIB coated IRE.60 The cooled ATR method was developed for VOCs and semi-volatile compounds analysis, improving the frequency of analysis and detection limits, around a few hundred μg L−1. ZnO nanomaterials have been used as IRE coatings, due to their high surface-to-volume ratio and specific interactions with organic functional groups, in order to improve the sensitivity and selectivity of VOC detection.61,62 ZnO nanowires provided a better performance for aromatic compounds while the ZnO nanospheres provided a better performance for polar VOCs with detection limits at ppm level.
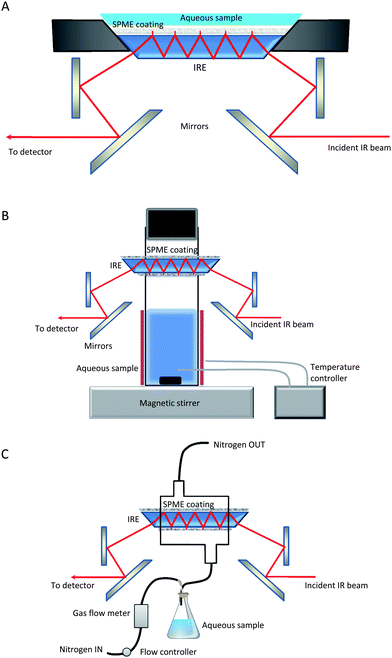
Purge-and-trap techniques have been largely applied to preconcentrate volatile compounds before analysis by gas chromatography or mass spectrometry and this technique has been also applied for the on-line extraction and preconcentration of VOCs from soil samples with ATR-FTIR spectroscopy. Vaporized semivolatile compounds, 1-chloronaphthalene, nitrobenzene and 2-chlorotoluene, were transported by a stream of nitrogen gas to the ATR flow cell.63 To increase the trapping efficiency, the IRE was coated with polyisobutylene, providing limits of detection of the order of 1 mg L−1. A similar approach was used for the determination of trichloroethylene, nitrobenzene and caffeine from aqueous solutions64 with detection limits of the order of 5 mg L−1.
 | ||
| Fig. 3 Manifold proposed for HS sampling LLME and ATR measurement in FTIR determination of VOCs in solid and liquid samples. | ||
The main limitation of the technique is that it could be only applied to volatile compounds and important interferences can be found in samples with high water content. To solve those problems, a new technique based on the combination of hydrodistillation and liquid phase microextraction was developed.66 The hydrodistillation step improved the extraction of volatile and semivolatile compounds and the liquid extraction using a chlorinated solvent, such as tetrachloroethylene, fulfills required conditions; such as: i) do not evaporate under the extraction conditions, ii) have the ability to extract the analytes efficiently, iii) be immiscible with water and iv) have a density higher than water in order to improve the contact of the vaporized analytes with the acceptor solvent and the ATR crystal.
The determination of pyrethroid and organochlorine pesticides in fresh water has been performed by combination of a LLE with n-hexane, followed by automated preconcentration by SPE using a silica mini-column, elution of the analyte with ethyl acetate and concentration and drying of the extract on the ATR element for spectrum acquisition.68 Recoveries for dichlofuanid, captan and fenpropathrin, from fortified tap and river waters, ranged from 66.3 to 102.0%, providing limits of detection of the order of 1.2 to 4.8 μg L−1.
Although the combination of different procedures provides lower detection limits than the use of a single one, complex methodologies become tedious and with a high sample handling, which reduces dramatically its applicability to real situations.
Powerful IR sources
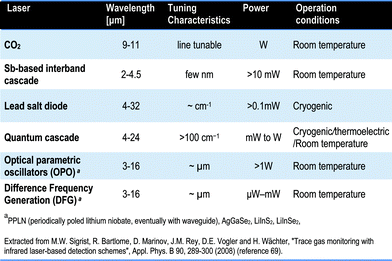
IR sources commonly employed include blackbody radiators, tungsten lamps, silicon carbide and other substances. In addition to these radiant sources, IR lasers emitting IR energy of a specific wavelength have also been used. For sensitivity reasons, coherent light sources emitting infrared radiation in the 2–12 μm range (MIR) are of considerable interest for many analytical applications in environmental monitoring and pollution control,69 detection of explosives70 and non-invasive disease diagnosis and therapy through breath analysis.71For quantitative IR measurements, the ideal laser source should have the following properties: i) sufficient optical power to ensure appropriate signal to noise ratios, ii) narrow linewidth to improve selectivity and sensitivity, iii) ease of tailoring the inherent laser operation wavelength, iv) low source noise, v) rapid wavelength tunability and vi) robust performance against changes in temperature, pressure, humidity and so on. IR laser spectroscopy has been used for years for detecting gas molecules in the mid-IR spectral region72 but also for big organic molecule analysis.73 The technique offers the advantages of high sensitivity, selectivity and fast time response. Promising mid-IR laser sources include CO2 lasers, lead-salt diode lasers, Antimonide-based laser diodes, quantum-cascade lasers (QCLs) in the mid-IR range above 4 μm, difference frequency generation (DFG) sources and optical parametric oscillators (OPOs).
Table 2 shows a comparison of the properties of the commonly used lasers in the MIR range in terms of wavelength, tenability, power and operation mode. For a more complete review on the solid state MIR sources, principles and applications, the reader should refer to specialized reviews and books.74,75,76
For instance, a photoacoustic system for the sensitive detection of ethene (C2H4) with a minimum detectable concentration of 6 ppt has been designed.79 Additionally, another procedure for NH3 monitoring reported a detection limit of 2–3 ppbV in ambient air.80 Moreover, 12 trace gases were measured simultaneously, mainly VOCs, in collected samples from car exhausts with the CO laser PA system.81
Although, tunable infrared laser differential absorption spectroscopy (TILDAS) with lead salt tunable diode lasers provides high sensitivity and selectivity for detecting gaseous molecules, the requirement of cryogenic cooling for lead salt lasers makes them inconvenient to use in many analytical chemistry applications. QCLs provide near-room-temperature operation with minimal wavelength drift.92 The application of IR laser spectroscopy to the quantification of glucose in aqueous solutions was investigated using cryogenically cooled lead salt lasers and QCLs operating at room temperature as IR sources. Fiber-based ATR spectroscopy and fiber-based transmission spectroscopy were employed as sampling modes, achieving detection limits of the order of 100 mg L−1.93 Detection of water in tetrahydrofurane with a limit of detection of 0.85 mg L−1 was achieved using a thermoelectrically cooled, pulsed, Fabry-Perot QCL, operating at 5.629 μm at room temperature.94 On-line monitoring of a model reaction, oxidation of sulfite to sulfate by hydrogen peroxide, was performed by employing two pulsed mid-infrared Fabry-Perot QCLs. The emission maxima of the QCLs were located at 1393 and 1080 cm−1 and the developed system allowed to monitor the reaction in the measurement cell with two QCLs operated simultaneously.95 QCLs have so far been used in numerous spectroscopic studies for environmental purposes, health monitoring, and military applications with detection limits of the order of parts per billion. Of course, distributed feedback QCLs are also suitable to long open path monitoring of ozone, water vapor and CO296 obtaining detection limits for ozone of the order of 0.3 ppm and real-time cigarette smoke analysis for ammonia (NH3), ethylene (C2H4), and nitric oxide (NO) concentrations.97
Surface enhanced infrared absorption (SEIRA) spectroscopy
The enhancement effect of the infrared spectra of molecules adsorbed on gold or silver island films is called surface enhanced infrared absorption (SEIRA).103 The strength of the enhancement depends on the metal type, on the metal film morphology because of the relation to the excitation spectrum of surface plasmons in the IR range and the analyte adsorbed.104The SEIRA effect could provide significant improvement of the sensitivity of IR measurements, its application in the chemical sensor field being a serious advantage. In the paper of H.D. Wanzenböck et al.105 an interesting question was raised: “whether the SEIRA principle can be applied to a variety of chemical substances regardless of their chemical structure?” It was concluded that a surface enhancement effect could be measured for aromatics, heteroaromatics and aliphatics with numerous different substituents. The increased absorption was observed for several functional groups such as amino-groups, chlorides, carboxylic acids, nitro-groups and sulfoxy-groups. In this paper, a linear correlation between the concentration and signal intensity was obtained for p-nitrobenzoic acid with an analyte surface coverage below 1700 ng corresponding to 10 nmol analyte. From those results, it can be concluded that the detection limit for a variety of analytes can be significantly lowered by using SEIRA.
The linkage of SEIRA with the sensor concept was achieved by R. Kellner et al. in 1997.106 Chemical interactions between the silver-island layer and the analytes (p-nitrobenzoic acid, and selected aromatic and chlorinated pesticides) proceed in a reversible manner which is an essential condition for the use of SEIRA in flow-through systems. An improvement of the IR intensity by a factor of 50 was achieved.
The main problem of SEIRA spectroscopy is that related to the preparation of SEIRA substrates by physical vapor deposition which is an expensive high vacuum method requiring precise and extensive control of all deposition parameters. The electrochemical production of SEIRA substrates was proposed to avoid those problems.107 The main advantages of this method are that no vacuum system is required, the fabrication of SEIRA-substrates is fast, the pretreatment of substrates is reduced and low-cost substrates can be prepared.
However, despite of the theoretical advantages offered by SEIRA, no practically applicable SEIRA sensor has been developed up to now.
Conclusions and future trends
As it can be seen in the previous sections the available tools for improving the sensitivity of IR determinations range from the improvements in instrumentation to efforts in sample preparation and thus it is clear that in the next few years IR will move from being the best technique for major and minor component analysis to also being a suitable technique for trace compounds.On comparing the advances on instrumentation and sample preparation it is clear that the enhancement of the obtainable limit of detection values strongly depend on the signal to noise ratio provided by the available instrument and this, the generalization of Fourier transform instruments, the improvement in detector systems from termopar and thermistors to DTGS and MCT and Peltier cooled systems, together with the extension of laser sources, are some of the milestones which will guarantee the future improvement of IR as a trace analysis technique. However, in our own opinion, the analytical effort in sample uptake, sample preconcentrations and measurement provide the main strategies for improving the analytical figures of merit independently on the instrumentation available and thus, additional effort in this direction will be of a general interest. In this sense, the future trends in trace analysis IR by incorporating sample preparation will came from the whole automation of the different steps which will permit the on-line combination of strategies also minimizing risks of errors and operator efforts and could provide validated procedures for sample analysis.
On the other hand a field which needs to be improved in the future is the search for general well integrated methodologies suitable for multicomponent trace analysis and in this way the efforts in the chemometric treatment of signals is mandatory to avoid the development of complex strategies to analyze single components.
In short, trace analysis by IR is a hot topic of research in today's analytical chemistry and additional efforts are required in different directions.
References
- A. L. Bluhm, J. A. Sousa and J. Weinstein, Appl. Spectrosc., 1964, 18, 188 CrossRef CAS.
- P. Norton, Opto-electronics Review, 2002, 10, 159 Search PubMed.
- J. U. White, J. Opt. Soc. Am., 1942, 32, 285 CrossRef.
- D. Herriott, H. Kogelnik and R. Kompfner, Appl. Opt., 1964, 3, 523 CrossRef.
- J. M. Garrigues, Z. Bouhsain, S. Garrigues and M. de la Guardia, Fresenius J. Anal. Chem., 2000, 366, 319 CrossRef CAS.
- J. Ohnsmann, G. Quintás, S. Garrigues and M. de la Guardia, Anal. Bioanal. Chem., 2002, 374, 561 CrossRef CAS.
- N. Mashkouri Najafi, A. Seyed Hamid and R. Khorrami Afshin, Microchem. J., 2003, 75, 151 CrossRef CAS.
- Z. Bouhsain, J. M. Garrigues, S. Garrigues and M. de la Guardia, Vib. Spectrosc., 1999, 21, 143 CrossRef CAS.
- M. Gallignani, M. Torres, C. Ayala and M. D. Brunetto, Revista técnica de la facultad de Ingeniería, Universidad de Zulia, 2008, 31, p. 159 Search PubMed.
- M. Gallignani, C. Ayala, M. R. Brunetto, J. L. Burguera and M. Burguera, Talanta, 2005, 68, 470 CrossRef CAS.
- R. Lucena, S. Cardenas, M. Gallego and M. Valcarcel, Anal. Chem., 2005, 77, 7472 CrossRef CAS.
- R. Lucena, S. Cardenas, M. Gallego and M. Valcarcel, Analyst, 2006, 131, 415 RSC.
- Y. Daghbouche, S. Garrigues, A. Morales-Rubio and M. de la Guardia, Anal. Chim. Acta, 1997, 345, 161 CrossRef CAS.
- N. El Hattab, Y. Daghbouche, M. El Hattab, L. Piovetti, S. Garrigues and M. de la Guardia, Talanta, 2006, 68, 1230 CrossRef CAS.
- N. Bouzidi, Y. Daghbouche, M. El Hattab, Z. Aliche, G. Culioli, L. Piovetti, S. Garrigues and M. de la Guardia, Anal. Chim. Acta, 2008, 616, 185 CrossRef CAS.
- M. T. Romero and N. Ferrer, Anal. Chim. Acta, 1999, 395, 77 CrossRef CAS.
- S. Garrigues, M. T. Vidal, M. Gallignani and M. de la Guardia, Analyst, 1994, 119, 659 RSC.
- Y. Daghbouche, S. Garrigues, M. T. Vidal and M. de la Guardia, Anal. Chem., 1997, 69, 1086 CrossRef CAS.
- Y. Daghbouche, S. Garrigues and M. de la Guardia, Anal. Chim. Acta, 1995, 314, 203 CrossRef CAS.
- H. LeThanh and B. Lendl, Anal. Chim. Acta, 2000, 422, 63 CrossRef.
- M. C. Alcudia-Leon, R. Lucena, S. Cardenas and M. Valcarcel, Anal. Chem., 2008, 80, 1146 CrossRef CAS.
- P. A. Tarantilis, V. E. Troianou, C. S. Pappas, Y. S. Kotseridis and M. G. Polissiou, Food Chem., 2008, 111, 192 CrossRef CAS.
- J. He, L. E. Rodriguez-Saona and M. M. Giusti, J. Agric. Food Chem., 2007, 55, 4443 CrossRef CAS.
- M. C. Alcudia-Leon, R. Lucena, S. Cardenas and M. Valcarcel, Anal. Chem., 2009, 81, 1184 CrossRef CAS.
- N. Ferrer and M. T. Romero, Microchim. Acta, 2002, 140, 35 CrossRef CAS.
- K. Yoshimura, H. Waki and S. Ohashi, Talanta, 1976, 23, 449 CrossRef CAS.
- A. Edelmann and B. Lendl, J. Am. Chem. Soc., 2002, 124, 14741 CrossRef CAS.
- M. Haberkorn, P. Hinsmann and B. Lendl, Analyst, 2002, 127, 109 RSC.
- A. Perez-Ponce and B. Lendl, Appl. Spectrosc., 2000, 54, 676 CrossRef.
- S. Armenta and B. Lendl, Vib. Spectrosc., 2009, 51, 60 CrossRef CAS.
- D. Perez-Palacios, S. Armenta and B. Lendl, Appl. Spectrosc., 2009, 63, 1015 CrossRef CAS.
- Z. Zhang and J. Pawliszyn, Anal. Chem., 1993, 65, 1843 CrossRef CAS.
- H. Y. Erbil, A. L. Demirel, Y. Avci and O. Mert, Science, 2003, 299, 1377 CrossRef CAS.
- S. X. Tan, Q. D. Xie, X. Y. Lu, N. Zhao, X. L. Zhang and J. Xu, J. Colloid Interface Sci., 2008, 322, 1 CrossRef CAS.
- M. Karlowatz, M. Kraft and B. Mizaikoff, Anal. Chem., 2004, 76, 2643 CrossRef CAS.
- G. Roy and J. A. Mielczarski, Water Res., 2002, 36, 1902 CrossRef CAS.
- F. Regan, F. Walsh and J. Walsh, Int. J. Environ. Anal. Chem., 2003, 83, 621 CrossRef CAS.
- A. M. S. Silva, M. F. Pimentel, I. M. Raimundo Jr. and Y. M. B. Almeida, Sens. Actuators, B, 2009, 139, 222 CrossRef.
- K. Flavin, H. Hughes and P. McLoughlin, Int. J. Environ. Anal. Chem., 2007, 87, 29 CrossRef CAS.
- J. Yang and Y. S. Huang, Appl. Spectrosc., 2000, 54, 202 CrossRef CAS.
- J. Yang and H. C. Lin, Analyst, 2000, 125, 1605 RSC.
- P. Heinrich, R. Wyzgol, B. Schrader, A. Hatzilazaru and D. W. Lubbers, Appl. Spectrosc., 1990, 44, 1641 CAS.
- R. Gobel, R. Krska, R. Kellner, R. W. Seitz and S. A. Tomellini, Appl. Spectrosc., 1994, 48, 678.
- J. Yang and M. L. Cheng, Analyst, 2001, 126, 881 RSC.
- J. Yang and C. P. Tsui, Anal. Chim. Acta, 2001, 442, 267 CrossRef CAS.
- G. T. Dobbs, B. Balu, C. Young, C. Kranz, D. W. Hess and B. Mizaikoff, Anal. Chem., 2007, 79, 9566 CrossRef CAS.
- B. Pejcic, M. Myers and A. Ross, Sensors, 2009, 9, 6232 CrossRef CAS.
- B. Mizaikoff, Anal. Chem., 2003, 75, 258A CAS.
- R. Krska, R. Kellner, U. Schiessl, M. Tacke and A. Katzir, Appl. Phys. Lett., 1993, 63, 1868 CrossRef CAS.
- M. Jakusch, B. Mizaikoff, R. Kellner and A. Katzir, Sens. Actuators, B, 1997, 38, 83 CrossRef.
- K. Flavin, J. Mullowney, B. Murphy, E. Owens, P. Kirwan, K. Murphy, H. Hughes and P. McLoughlin, Analyst, 2007, 132, 224 RSC.
- J. Yang, H. J. Lin and H. Y. Huang, Anal. Chim. Acta, 2005, 530, 213 CrossRef CAS.
- J. S. Lee and J. Yang, Anal. Biochem., 2006, 359, 124 CrossRef CAS.
- Y. K. Wei and J. Yang, Talanta, 2007, 71, 2007 CrossRef CAS.
- H. C. Lin, Y. H. Chou and J. Yang, Anal. Chim. Acta, 2008, 611, 89 CrossRef CAS.
- B. McAuley and S. E. Cabaniss, Anal. Chim. Acta, 2007, 581, 309 CrossRef CAS.
- J. Yang and J. W. Her, Anal. Chem., 2000, 72, 878 CrossRef CAS.
- J. Yang and S. S. Tsai, Anal. Chim. Acta, 2001, 436, 31 CrossRef CAS.
- J. Yang and W. C. Chen, Int. J. Environ. Anal. Chem., 2004, 84, 1045 CrossRef CAS.
- J. Yang and S. S. Tsai, Anal. Chim. Acta, 2002, 462, 235 CAS.
- G. G. Huang, C. T. Wang, H. T. Tang, Y. S. Huang and J. Yang, Anal. Chem., 2006, 78, 2397 CrossRef CAS.
- J. Yang, Y. R. Shih, I. C. Chen, C. I. Kuo and Y. S. Huang, Appl. Spectrosc., 2005, 59, 1002 CrossRef CAS.
- J. Yang and J. W. Her, Anal. Chem., 1999, 71, 4690 CrossRef CAS.
- J. Yang and J. W. Her, Anal. Chem., 1999, 71, 1773 CrossRef CAS.
- A. Gonzalvez, S. Garrigues, S. Armenta and M. de la Guardia, Anal. Chem., 2010, 82, 3045 CrossRef CAS.
- A. Gonzalvez, S. Garrigues, S. Armenta and M. de la Guardia, Anal. Bioanal. Chem., in press DOI:10.1007/s00216-010-4020-1.
- K. Kargosha, S. H. Ahmadi, A. Ghassempour and M. R. Arshadi, Analyst, 1999, 124, 367 RSC.
- A. Colume, J. Diewok and B. Lendl, Int. J. Environ. Anal. Chem., 2004, 84, 835 CrossRef CAS.
- M. W. Sigrist, R. Bartlome, D. Marinov, J. M. Rey, D. E. Vogler and H. Wachter, Appl. Phys. B: Lasers Opt., 2008, 90, 289 CrossRef CAS.
- C. Bauer, A. K. Sharma, U. Willer, J. Burgmeier, B. Braunschweig, W. Schade, S. Blaser, L. Hvozdara, A. Müller and G. Holl, Appl. Phys. B: Lasers Opt., 2008, 92, 327 CrossRef CAS.
- C. Wang and P. Sahay, Sensors, 2009, 9, 8230 CrossRef CAS.
- M. Zahniser, D. Nelson, C. Kolb, in: K. Kohse-Hoinghaus, J. Jeffries (ed.), Applied Combustion Diagnostics, Taylor and Francis, New York, 2002 (Chapter 26) Search PubMed.
- R. Bartlome, J. M. Rey and M. W. Sigrist, Anal. Chem., 2008, 80, 5334 CrossRef CAS.
- A. Godard, C. R. Phys., 2007, 8, 1100 Search PubMed.
- R. F. Curl and F. K. Tittel, Annu. Rep. Prog. Chem., Sect. C, 2002, 98, 219 RSC.
- I. T. Sorokina and K. L. Vodopyanov (ed.), Solid-State Mid-Infrared Sources, Topics in Applied Physics, vol. 89, Springer, Berlin, Heidelberg, 2003 Search PubMed.
- M. W. Sigrist and A. Thoeny, Proc. SPIEVol. 1715, p. 174–184, Optical Methods in Atmospheric Chemistry, Harold I. Schiff; Ulrich Platt; Eds, 1993 Search PubMed.
- A. Romann and M. W. Sigrist, Appl. Phys. B: Lasers Opt., 2002, 75, 377 CrossRef CAS.
- F. J. M. Harren, J. Reuss, E. J. Woltering and D. D. Bicanic, Appl. Spectrosc., 1990, 44, 1360 CAS.
- H. Sauren, E. Gerkema, D. Bicanic and H. Jalink, Atmos. Environ., 1993, 27A, 109 CrossRef CAS.
- S. Bernegger and M. W. Sigrist, Infrared Phys., 1990, 30, 375 CrossRef.
- M. Tacke, Infrared Phys. Technol., 1995, 36, 447 CrossRef CAS.
- M. Feher and P. A. Martin, Spectrochim. Acta, Part A, 1995, 51, 1579 CrossRef.
- P. Werle, Spectrochim. Acta, Part A, 1998, 54, 197 CrossRef.
- A. Joullié, P. Christol, A. N. Baranov and A. Vicet, Mid-infrared 2–5 μm heterojunction laser diodes, in: I. T. Sorokina and K. L. Vodopyanov (ed.), Solid-State Mid-Infrared Sources, in: Topics in Applied Physics, vol. 89, Springer, Berlin, Heidelberg, 2003, pp. 1–59 Search PubMed.
- A. Vicet, D. A. Yarekha, A. Pérona, Y. Rouillard, S. Gaillard and A. N. Baranov, Spectrochim. Acta, Part A, 2002, 58, 2405 CrossRef CAS.
- D. Z. Garbuzov, H. Lee, V. Khalfin, R. Martinelli, J. C. Connolly and G. L. Belenky, IEEE Photonics Technol. Lett., 1999, 11, 794 CrossRef.
- E. Geerlings, M. Rattunde, J. Schmitz, G. Kaufel, H. Zappe and J. Wagner, IEEE Photonics Technol. Lett., 2006, 18, 1913 CrossRef CAS.
- M. Hümmer, K. Rößner, A. Benkert and A. Forchel, IEEE Photonics Technol. Lett., 2004, 16, 380 CrossRef.
- P. Werle and A. Popov, Appl. Opt., 1999, 38, 1494 CrossRef CAS.
- O. Malis, C. Gmachl, D. L. Sivco, L. N. Pfeiffer, A. M. Sergent and K. W. West, AT&T Bell Lab. Tech. J., 2005, 10, 199 Search PubMed.
- F. Capasso, C. Gmachl, D. L. Sivco and A. Y. Cho, Phys. Today, 2002, 55, 34–38 CrossRef CAS.
- A. Lambrecht, T. Beyer, K. Hebestreit, R. Mischler and W. Petrich, Appl. Spectrosc., 2006, 60, 729 CrossRef CAS.
- A. Ouvrard, K. O'Dwyer and B. D. MacCraith, Appl. Spectrosc., 2008, 62, 1349 CrossRef CAS.
- S. Schaden, A. Dominguez-Vidal and B. Lendl, Appl. Spectrosc., 2006, 60, 568 CrossRef CAS.
- M. Taslakov, V. Simeonov and H. van den Bergh, Spectrochim. Acta, Part A, 2006, 63, 1002 CrossRef CAS.
- Q. Shi, D. D. Nelson, J. B. McManus, M. S. Zahniser, M. E. Parrish, R. E. Baren, K. H. Shafer and C. N. Harward, Anal. Chem., 2003, 75, 5180 CrossRef CAS.
- W. Chen, J. Cousin, E. Poullet, J. Burie, D. Boucher, X. Gao, M. W. Sigrist and F. K. Tittel, C. R. Phys., 2007, 8, 1129 Search PubMed.
- D. Rehle, D. Leleux, M. Erdelyi, F. Tittel, M. Fraser and S. Friedfeld, Appl. Phys. B, 2001, 72, 947 CAS.
- A. Fried, G. Diskin, P. Weibring, D. Richter, J. G. Walega, G. Sachse, T. Slate, M. Rana and J. Podolske, Appl. Phys. B: Lasers Opt., 2008, 92, 409 CrossRef CAS.
- D. Richter, M. Erdelyi, R. F. Curl, F. K. Tittel, C. Oppenheimer, H. J. Duffell and M. Burton, Opt. Lasers Eng., 2002, 37, 171 CrossRef.
- C. Fischer, R. Bartlome and M. W. Sigrist, Photons plus Ultrasound: Imaging and Sensing II, Proc. SPIE, 2005, 5697, 56 CAS.
- A. Hatta, T. Ohshima and W. Suëtaka, Appl. Phys. A: Mater. Sci. Process., 1982, 29, 71 CrossRef.
- D. Ross and R. Aroca, J. Chem. Phys., 2002, 117, 8095 CrossRef CAS.
- H. D. Wanzenbok, B. Mizaikoff, N. Weissenbacher and R. Kellner, J. Mol. Struct., 1997, 410–411, 535.
- R. Kellner, B. Mizaikoff, M. Jakusch, H. D. Wanzenbock and N. Weissenbacher, Appl. Spectrosc., 1997, 51, 495 CrossRef CAS.
- H. D. Wanzenböck, B. Mizaikoff, N. Weissenbacher and R. Kellner, Fresenius J. Anal. Chem., 1998, 362, 15 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |