Electrochemiluminescence of 9,10-diphenylanthracene doped polystyrene beads in aqueous media
Guo
Guangmei
a,
Zhijie
Lin
b and
Xi
Chen
*b
aCollege of Sciences, Hebei University of Science and Technology, Shijiazhuang, 050018, China
bState Key Laboratory of Marine Environmental Science, Xiamen University, Xiamen, 361005, China. E-mail: xichen@xmu.edu.cn; Tel: +86-592-2184530
First published on 8th December 2010
Abstract
In order to use a water soluble organic electrochemiluminescence (ECL) reagent in an aqueous medium, a simple one-pot method was developed to synthesize 9,10-diphenylanthracene (9,10-DPA) doped polystyrene beads based on dispersion polymerization. The bead size was around 3 μm and they were well dispersed. Fluorescence experiments showed that the polystyrene beads were successfully doped in 9,10-DPA. The 9,10-DPA doped beads were then used in ECL studies, and they retained their ECL activity well. The results imply that polystyrene beads could be a suitable platform leading to the application of hydrophobic ECL reagents in aqueous media.
Since the first reports of tris(2,2′-bipyridyl)ruthenium(II) (Ru(bpy)32+) electrochemiluminescence (ECL),1–3Ru(bpy)32+ has become dominant in ECL studies because of its solubility and high ECL efficiency in aqueous solutions. As a result, Ru(bpy)32+ applications have been used in many fields.4–8 Especially after the development of a solid-state ECL technique9–13 which allows the reuse of Ru(bpy)32+, the applications of Ru(bpy)32+ have been greatly extended. However, the obvious weakness of the Ru(bpy)32+ based ECL system is that the single wavelength of Ru(bpy)32+-based ECL makes the analytical capacity of ECL analysis very limited.14 The combination with high performance liquid chromatography15,16 or electrophoresis chromatography17–19 is necessary, when a single sample contains several analytes. This makes the ECL analytical system very complicated. In order to resolve this problem, a great number of metal complexes and organic compounds have been synthesized.4–8 Some of the compounds present ECL efficiency comparable to that of Ru(bpy)32+ in organic solvents, for example 9,10-diphenylanthracene (DPA, whose ECL reaction routes with tripropylamine are shown in Scheme 120). However, most of the compounds are unavailable in aqueous solution due to their low solubility. DPA has a carboxylic group which increases its solubility in aqueous solution, but the result is not very satisfactory,21 and so the application of organic ECL reagents in aqueous solution remains a problem. It is reported that Ru(bpy)32+ can be concentrated in silica nanoparticles with extremely high ECL ability using a microemulsion method.22ECL reagent immobilized nanoparticles could be an effective approach to resolve the problem mentioned above. In this paper, we developed a simple one-pot synthesis method for doping DPA in polystyrene beads (DPA@PS) in order to study its ECL. The DPA@PS ECL results indicated that polystyrene (PS) beads could be a suitable platform leading to the application of hydrophobic ECL reagents in aqueous media.
 | ||
| Scheme 1 ECL reaction processes of TPA and DPA | ||
9,10-Diphenylanthracene (9,10-DPA, 97%), 5 wt.% Nafion perfluorinated ion-exchange resin (Nafion), tripropylamine (TPA, 99%), styrene (99%) and methacrylic acid (MAA, 99%) were purchased from Sigma-Aldrich (USA). Polyvinylpyrrolidone K30 (PVP) and azobisisobutyronitrile (AIBN) were purchased from Shanghai National Chemical Company (China). All solutions were prepared with ultrapure water obtained from a Millipore autopure WR600A system (USA). Square glassy carbon plates (GCPs, 1 cm × 1 cm × 0.1 cm, L × W × H) were purchased from BAS Co. Ltd., (Japan).
70 mg 9,10-DPA was dissolved in 70 mL styrene. Successively, 0.0625 g AIBN and 70 μL MAA were added to the solution with magnetic stirring in the dark and deoxygenized with N2 flow. A deoxygenized solution containing 0.1875 g PVP and 23.75 mL anhydrous ethanol was then added. The mixture was sealed and placed in a 70 °C oil-bath with magnetic stirring for 24 h. The resulting PS beads were centrifuged at 8000 rpm, washed five times with ethanol and then dried at 4 °C.
A GCP was polished with 0.3 μm, 0.05 μm Al2O3 powder before use, and then dipped into 20 μL 0.5 wt.% Nafion ethanol solution containing 25 mg mL−1 DPA@PS. The modified GCP was then dried in the dark at room temperature. Next, a current conducting wire was adhered to the GCP, which was then sealed using waterproof adhesive tape to make a glassy carbon electrode (GCE). The working area of the DPA@PS@GCE was 70 mm2.
ECL experiments were carried out with a three-electrode system using a DPA@PS@GCE as the working electrode, a saturated calomel electrode (SCE) as the reference electrode, and a platinum wire as the counter electrode. Potential control was achieved using a CHI 660b electrochemical working station (Chenghua Co. Ltd., Shanghai, China). The ECL intensity was detected using an IFFM-D flow-injection system (Ruimai Co. Ltd., Xi'an, China) with high voltage at −900 V. Fluorescence spectra were obtained using a Hitachi F-4500 fluorescence spectrophotometer (Hitachi Co. Ltd., Japan). A scanning electron microscope (SEM; LEO1530, Leo, Germany) was used to obtain scanning electron micrographs of the DPA@PS.
Because DPA has many phenyl groups, this causes it to be insoluble in aqueous solutions and difficult to be concentrated on silica nanoparticles as in prior report methods for Ru(bpy)32+ doping. In general, DPA dissolves well in phenyl-containing solvents such as styrene. In the synthesis of PS, styrene is used in the dispersion polymerization method. DPA is concentrated inside the PS beads when DPA is dissolved in styrene during the reaction. The SEM image (Fig. 1) indicates that the size of DPA@PS was around 3 μm and that the doped beads were well dispersed.
 | ||
| Fig. 1 SEM image of DPA@PS. | ||
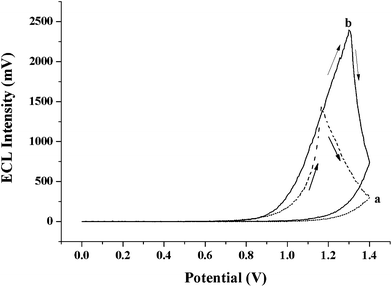
The DPA@PS generated ECL when the DPA@PS@GCE was immersed in 0.1 M phosphate buffer solution containing 1 mM TPA. As shown in Fig. 2, the ECL intensity was relatively strong and increased when the applied potential was above 0.8 V. It can also be seen that the ECL profile was delayed from the oxidation peak, since ECL was caused by the reaction between the radicals of TPA+ and DPA+ as shown in Scheme 1, and the oxidation potential of TPA was around 0.8 V i.e. prior to that of DPA. The results indicated that DPA retained its ECL ability as reported20,23 and could emit relatively strong ECL when it was doped in the PS beads. Using this approach, DPA could be concentrated at high concentration in the PS for ECL applications. Generally, the balance between electrode reaction and diffusion rate in an ECL process generates a maximum ECL signal value, and then the ECL value remains constant or decreases gradually. However, as shown in Fig. 3, sharply decreasing DPA ECL intensities (with/without TPA) were found after the applied potential exceeded 1.18 V and 1.32 V, respectively, which is different from the ECL spectra of other reagents in solution.4 The excitation period is ordinarily longer for fluorescent probes containing larger conjugate π bonds, resulting in more obvious quenching of luminescence. In addition, the shift in ECL peaks seen in Fig. 3 shows that the ECL peak at a potential of 1.18 V without TPA changed to 1.32 V with TPA. When the applied potential was selected at or above 1.0 V, the oxygen on the electrode surface, generated from the electrolysis of water, reacted with the TPA radical, which reduced the quenching effect of oxygen on DPA ECL. The ECL peak with TPA at 1.32 V might be contributed mainly by the DPA radical. Simultaneously, the ECL peak without TPA at 1.18 V might directly be from the reaction of the oxygen radical and DPA.
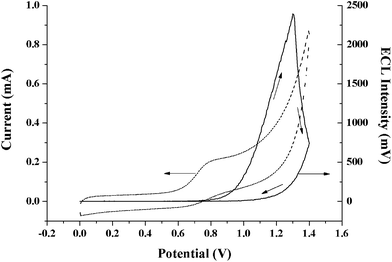 | ||
| Fig. 2 Constant voltage (dashed line) and ECL (solid line) spectra of DPA@PS@GCE in 0.1 M PBS (pH 9.0) containing 1 mM TPA. | ||
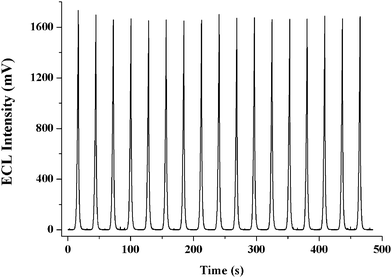
Since the ECL intensity of DPA@PS was affected by oxygen, further experiments were carried out to confirm the applicability of DPA@PS@GCE in ECL analysis. DPA@PS@GCE presented a good linear response to different concentrations of TPA, ranging from 10−3 to 10−7 M. The detection limit for TPA was found to be 50 nM. In addition, as shown in Fig. 4, the ECL intensity of DPA@PS@GCE was very stable during multi-potential scanning. The results indicated that DPA@PS@GCE was suitable for ECL analysis.
Conclusions
In order to apply organic ECL reagents in aqueous ECL analysis, a simple one-pot method was developed based on the dispersion polymerization method. DPA was selected as the ECL reagent, and was dissolved in styrene during the polymerization reaction. The resulting PS beads were examined using a fluorescence and ECL experiment. This experiment indicated that DPA was successfully doped in the PS beads and DPA@PS presented a good ECL performance with a linear range from 10−3 M to 10−7 M. The detection limit was found to be 50 nM for TPA. Although the ECL intensity was still affected by oxygen, PS might be a suitable platform to apply organic ECL reagents in aqueous solution. This approach will provide more choices in the application of hydrophobic ECL reagents, extending their use to aqueous media such as biological fluids.Acknowledgements
This research was financially supported by the National Natural Scientific Foundation of China (No. 20775064 and 20975085), which is gratefully acknowledged. Furthermore, we would like to extend our thanks to Professor John Hodgkiss of The University of Hong Kong for his assistance with English.Notes and references
- N. E. Tokel and A. J. Bard, J. Am. Chem. Soc., 1972, 94, 2862–2863 CrossRef CAS.
- M. Chang, T. Saji and A. J. Bard, J. Am. Chem. Soc., 1977, 99, 5399–5403 CrossRef CAS.
- I. Rubinstein and A. J. Bard, J. Am. Chem. Soc., 1981, 103, 512–516 CrossRef CAS.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS.
- D. Dini, Chem. Mater., 2005, 17, 1933–1945 CrossRef CAS.
- Y. Du and E. Wang, J. Sep. Sci., 2007, 30, 875–890 CrossRef CAS.
- J. F. Rusling, E. G. Hvastkovs, D. O. Hull and J. B. Schenkman, Chem. Commun., 2008, 141–154 RSC.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS.
- C. J. Miller, P. McCord and A. J. Bard, Langmuir, 1991, 7, 2781–2787 CrossRef CAS.
- I. Rubinstein and A. J. Bard, J. Am. Chem. Soc., 1980, 102, 6641–6642 CrossRef.
- Z. Guo and S. Dong, Anal. Chem., 2004, 76, 2683–2688 CrossRef CAS.
- Y. Du, B. Qi, X. Yang and E. Wang, J. Phys. Chem. B, 2006, 110, 21662–21666 CrossRef CAS.
- T. Downey and T. A. Nieman, Anal. Chem., 1992, 64, 261–268 CrossRef CAS.
- D. Bruce and M. M. Richter, Anal. Chem., 2002, 74, 1340–1342 CrossRef CAS.
- H. Morita and M. Konishi, Anal. Chem., 2002, 74, 1584–1589 CrossRef CAS.
- S. P. Forry and R. M. Wightman, Anal. Chem., 2002, 74, 528–532 CrossRef CAS.
- J. Liu, J. Yan, X. Yang and E. Wang, Anal. Chem., 2003, 75, 3637–3642 CrossRef CAS.
- X. Yin, H. Qiu, X. Sun, J. Liu and E. Wang, Anal. Chem., 2004, 76, 3846–3850 CrossRef CAS.
- J. Li, Q. Yan, Y. Gao and H. Ju, Anal. Chem., 2006, 78, 2694–2699 CrossRef CAS.
- K. S. V. Santhanam and A. J. Bard, J. Am. Chem. Soc., 1965, 87, 139–140 CrossRef CAS.
- T. C. Richards and A. J. Bard, Anal. Chem., 1995, 67, 3140–3147 CrossRef CAS.
- L. Zhang and S. Dong, Anal. Chem., 2006, 78, 5119–5123 CrossRef CAS.
- D. J. Vinyard, S. Su and M. M. Richter, J. Phys. Chem. A, 2008, 112, 8529–8533 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |