Ion/molecule reactions for detecting ammonia using miniature cylindrical ion trap mass spectrometers†
Jonell N.
Smith
a,
Adam D.
Keil
b,
Robert J.
Noll
*ac and
R. Graham
Cooks
ac
aDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA. E-mail: rnoll@purdue.edu; Fax: +1 765 494 9421; Tel: +1 765 494 5265
bIndependent Contractor, Monmouth, IL 61462, USA
cCenter for Analytical Instrumentation Development, Purdue University, Bindley Bioscience Center, Discovery Park, 1203 W. State St, West Lafayette, IN 47907, USA
First published on 25th October 2010
Abstract
Gaseous ammonia, a common toxic industrial compound, is not detected readily in ion trap mass spectrometers because its molecular ion falls below the low-mass cutoff (∼m/z 40) normally used when examining organic compounds. Instead, reactions of ammonia with halobenzene radical cations were used with internal electron ionization in two cylindrical ion trap miniature mass spectrometers to create a characteristic product ion by which to identify and quantify ammonia. Ammonia showed a linear response over the concentration range studied (parts per million [ppm] to parts per billion [ppb]) with limits of detection of 17 ppm and 220 ppb for experiments involving direct introduction and thermal desorption after pre-concentration, respectively. These values are comparable to ammonia's permissible exposure limit (50 ppm) and odor threshold (5 ppm). Receiver operating characteristic (ROC) curves were used to describe the method sensitivity, the probability of true positives, and the false positive rate for ammonia. A customized reaction scan function was created to select the species available for the ion/molecule reaction and set the amount of time the product ion could be accumulated in the trap. Product ion identity was verified using tandem mass spectrometry. Similar reactions with methylamine, ethylamine and the two nitriles, acetonitrile and benzonitrile, were explored.
Introduction
As the need for in situ analysis of low levels of analytes in complex matrices has increased, efforts have been made to miniaturize laboratory scale instrumentation. Analytical objectives include environmental monitoring, industrial process control, facility air monitoring, homeland and transportation security, and chemical spill monitoring. Mass spectrometry (MS) has been one area of focus in terms of miniaturization of analytical instrumentation1 because of the accuracy, sensitivity, and selectivity it has to offer for a variety of biological and chemical species. Mass spectrometers with a large mass-to-charge (m/z) range are needed for many of these applications; mass analyzers are typically not optimized for very low m/z ions and switching between high- and low-mass ranges can be slow and inconvenient.Analytes whose characteristic ions have low m/z ratios (<m/z 50) are typically poorly detected using miniature mass spectrometers. This is because most such instruments are based on quadrupole ion traps and there is an inverse quadratic relationship between mass and internal radius of the trap.2 The addition of a reagent (either neutral or an ionized molecule) to react with the small analyte molecule within the ion trap, thereby producing a product ion of higher m/z, is a possible approach to improving sensitivity, detection limits, and overall quantitative performance with respect to that analyte. Furthermore, in the ideal case, sufficiently complex product ions will be formed so that collision-induced dissociation (CID) will produce new characteristic ions for the purposes of secure analyte identification through multiple stage mass spectrometry (MSn). Small ions (e.g., m/z 18 NH4+) are often not well-suited for MSn experiments since they fragment poorly and/or uncharacteristically. Though ion/molecule reactions have been studied in the past using miniature mass spectrometers,3–5 we report here, to the best of our knowledge, the first approach in which an ion/molecule reaction has been used for the detection of a low-mass analyte on a miniaturize mass spectrometer.
The toxic industrial compound ammonia (NH3) has been described by the US National Institute for Occupation Safety and Health (NIOSH) as a colorless gas with a pungent, suffocating odor that can cause damage to the eyes, skin, and respiratory system under certain conditions. The immediate danger to life and health (IDLH) level is 300 parts per million (ppm), the permissible exposure limit (PEL) is 50 ppm, and the odor threshold is 5 ppm.6 In 2006, worldwide production of ammonia for uses such as household cleaners, fertilizers, a food production antimicrobial agent, industrial refrigeration, and explosives was estimated to be 146.5 million tons.7 On-site monitoring of this chemical's release with a portable mass spectrometer thus has potential value in many areas.
Ammonia has a high proton affinity (PA) of 853.6 kJ mol−18 so it readily forms the protonated ammonium ion (m/z 18 NH4+) in positive ion mode MS. Both this ion and the protonated dimer (m/z 35) fall below the low-mass cutoff (LMCO, ∼m/z 40) of a typical miniature mass spectrometer4 under normal operating conditions. For the same reason, positive ion mode atmospheric pressure ion/molecule reactions were used by Cotte-Rodríguez et al. on a commercialized Finnigan LTQ linear ion trap mass spectrometer to detect ammonia (5 to 50 ppm) via an ion/molecule reaction with carbon dioxide (CO2).9 Two adduct ions of interest [m/z 61 (CO2·NH3)+ and m/z 124 (2CO2·H2O·NH4)+] were detected and the identity of the ion of m/z 124 was confirmed with MS3 experiments. Calibration curve parameters were reported (y = 36.983x + 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 597, R2 = 0.9639, RSD < 3%) and the response was linear over an ammonia concentration range of 5 to 50 ppm. Taking this modest performance into account, we investigated a different ion/molecule reaction involving a liquid reagent that could perhaps be implemented in a fieldable instrument more easily than a canister of CO2.
597, R2 = 0.9639, RSD < 3%) and the response was linear over an ammonia concentration range of 5 to 50 ppm. Taking this modest performance into account, we investigated a different ion/molecule reaction involving a liquid reagent that could perhaps be implemented in a fieldable instrument more easily than a canister of CO2.
Ion/molecule reactions of ammonia,10–16 including those in which ammonia is used as a chemical ionization (CI) gas,17 can generate products of higher m/z values. However, many of these reactions yield multiple products in small quantities, and some of these products are below the desired mass range (≥m/z 50). Thölmann and Grützmacher reported on nucleophilic aromatic substitution reactions of halobenzene radical cations with ammonia in the gas phase.18 These reactions yielded product ions with (i) mass/charge much greater than our LMCO value, (ii) in large intensity, and (iii) unique fragmentation behavior in MSn experiments to serve as a good structural confirmation tool. This suggests that halobenzenes are viable reagents for ammonia determination.
This reaction involves the preparation of the halobenzene radical cation as a reagent ion using electron impact (EI) ionization and a substitution reaction with neutral ammonia gas to give the anilinium ion derivative (reaction (1)). Gaseous ammonia was introduced into either of two miniature mass spectrometers (Griffin 300 and ChemSense 600) either directly into the vacuum chamber or onto a solid sorbent material, with subsequent thermal desorption of the condensable vapors for internal EI ionization and mass analysis. Five halobenzenes (bromobenzene, bromobenzene-d5, chlorobenzene, 1,2-dichlorobenzene, and 1,2,4-trichlorobenzene) were investigated for suitability of the main reaction (1), while nitriles (acetonitrile and benzonitrile) and two amines (methylamine and ethylamine) were also investigated in similar reactions. The reaction pathway was evaluated. Reaction of molecular ions of halogenated benzenes proved useful for the detection, identification, and quantification of ammonia in the gas phase.
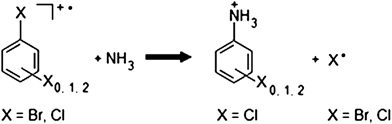 | (1) |
Experimental
Instrumentation
Two miniature cylindrical ion trap (CIT) mass spectrometers (Griffin 300, Griffin System Software (GSS) v. 3.4, and ChemSense 600, GSS v. 3.6, ICx Analytical Instruments, West Lafayette, IN) were used in the positive ion mode for all experiments. Operating pressure of both instruments was <1 × 10−5 Torr. The Griffin 300 is similar to previously described custom instrumentation.5,19–21 It has a direct leak inlet system. Electrons for EI ionization are generated by thermionic emission from a rhenium filament. The ChemSense 600 uses a glow discharge EI (GDEI) ionization source22,23 to produce electrons, but produces similar mass spectra.24 The ChemSense 600, recently described for use in automated facility air monitoring,24 was operated in the manual mode using either its direct leak inlet or a single sorbent preconcentration tube (Tenax TA/Carboxen 569, 3 mm diameter × 86 mm length, 25 mg each, Chemical Agent Monitoring Supply Co., Houston, TX, heated to 150 °C).The scan function (Fig. 1) consisted of six periods: (1) reagent ion preparation by EI ionization (0.05 to 150 ms), duration controlled by automatic level control (ALC) algorithm; (2) cooling (10 ms); (3) ramp RF amplitude (10 ms) and (4) hold RF amplitude (50 ms) to eject lower m/z fragment ions created during the preparation of the reagent ions; the duration of the RF hold represents a compromise between short times which favor reagent cation signal intensity and long times which favor more complete low m/z fragment ion ejection. Ions at m/z > reagent ion were not observed in these experiments, so there was no need to eject at higher m/z. (5) Product ion accumulation time (PIAT, up to 1000 ms), the LMCO was set to m/z 50 trapping reaction products with m/z > 50. (6) Mass analysis (20 ms), ramping the RF amplitude (Griffin 300: RF = 1.5 MHz; ChemSense 600: RF = 1.6 MHz) in the mass-selective instability scan with resonant ejection (AC = 360 kHz and 373 kHz, respectively) with ions detected with an electron multiplier. An average of 10 scans was used to create each mass spectrum unless otherwise stated.
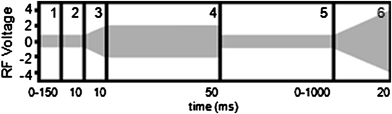 | ||
| Fig. 1 Graphical depiction of the custom reaction scan function for low-mass ejection before reaction of the reagent molecular cations with ammonia. The segments are: (1) ionization: the time period for this step was variable and controlled by the software's ALC algorithm, (2) ion cooling, (3) RF ramp to RF level in next section, (4) RF level such that the effective LMCO was slightly below the m/z of the ions to be isolated (i.e., low-mass ejection), (5) LMCO returned to m/z 50, the product ion accumulation time segment, and (6) RF ramp for mass analysis from m/z 50 to 300. | ||
Chemicals and standards
All chemicals were commercially available and used without further purification. A high-pressure cylinder of ammonia (503 ppm, nitrogen as balance gas, Air Liquide America Specialty Gases LLC, Plumsteadville, PA) was used both at full concentration and also diluted using OSHA D breathable compressed air (Indiana Oxygen, Indianapolis, IN). Perfluorotributylamine (PFTBA, Aldrich) headspace vapor was used as a mass calibrant for both instruments. All other reagents were obtained from Sigma-Aldrich (Milwaukee, WI).Sample preparation and introduction system
The sample preparation and the sample introduction systems are described briefly here but in full detail in the ESI; see Fig. S1† for a diagram. Gaseous ammonia was introduced using a gas-phase sample introduction system composed of two mass flow controllers (MFCs; MKS Instruments, Andover, MA; Teledyne Hastings Instruments, Hampton, VA) fitted with Teflon tubing (0.6 cm diameter; Swagelok, Indianapolis, IN) and stainless steel Swagelok fittings. Dilutions with OSHA D breathable compressed air produce analyte concentrations ranging from low-ppm to the maximum concentration of the ammonia cylinder (503 ppm).The relative reaction rates of ammonia, methylamine, and ethylamine with 1,2-dichlorobenzene were studied. Neat liquid methylamine and ethylamine were delivered separately via syringe (Hastings 81165, 250 µL, 3.26 mm diameter) at a heated (∼90 °C) Teflon tee (Swagelok) into a steady stream of the ammonia gas standard using a syringe pump (Harvard Apparatus) such that ammonia and the two amines were delivered to the instrument at a concentration of 503 ppm.
An external roughing pump was used to evacuate atmospheric air from the Griffin 300's vacuum system before performing a direct air leak experiment. A piece of PEEKsil (length 10 cm, inner diameter 50 µm, SGE, Inc., Austin, TX) capillary was used to insure a constant, restricted flow (0.25 mL min−1) of the analyte gas stream to the stainless steel union at which headspace vapors above a neutral reagent compound were introduced by leak valve (Granville-Phillips, Series 203). When using the ChemSense 600, headspace vapor of the reagent was introduced directly to the vacuum chamber.
In some experiments the ChemSense 600's dual sorbent tube sample inlet system24 was used to pre-concentrate low gas phase concentrations of ammonia. Sample was collected onto the sorbent tube bedding for 30 s. After 3 s, the mass spectrometer scan was begun using the customized reaction scan function, as described in detail below. During the next 40 seconds, the mass spectrometer was continuously scanned while residual air in the tube was evacuated (10 s), sample was desorbed from the sorbent material by heating (150 °C for 20 s), and the heater was turned off so that the tube could cool (10 s) for the remainder of the mass scan period. The sorbent tube was thus interrogated for a total of 73 s.
In the special case where acetonitrile, benzonitrile, and aqueous ammonia headspace were introduced, sampling took place directly at the PEEKsil capillary on the Griffin 300 or directly at the sample inlet on the ChemSense 600.
Quantitative analysis
Reconstructed ion chronograms (RICs) are plots of the signal intensity for ions of a particular m/z value or range of m/z values versus time. Instrument response was taken as the integrated RIC area for a specified peak using a baseline of zero over a specified period of time. LODs were obtained with 95% confidence using a method that takes into account degrees of freedom, as laid out by Currie,25 and used and described in detail by us in the past.24Results and discussion
Halobenzenes as reagents in ion/molecule reactions with ammonia
As depicted in reaction (1), halobenzene radical cations, created using internal EI ionization, when reacted with neutral ammonia, produce haloanilinium ions and a halogen radical. Five halobenzenes were investigated in this study for their suitability for the quantitative analysis of ammonia: bromobenzene, bromobenzene-d5, chlorobenzene, 1,2-dichlorobenzene, and 1,2,4-trichlorobenzene. Single-stage mass spectra were recorded for all five halobenzenes, as well as spectra for the ammonia reaction (see ESI, Fig. S2–S6†). Bromobenzene-d5 was used to confirm our results, where the ammonia reaction product was five m/z units greater than that of bromobenzene with ammonia, as expected. Vapor pressure,26 reagent ion formula and m/z, product ion formula and m/z, reaction efficiency18 when available, and other ions of interest detected in individual spectrum and their m/z are summarized in Table 1. All products arose as expected from reaction (1).| Reagent precursor | Vapor pressurea (25 °C)/mbar | Reagent ion formula (m/z) | Product ion formula (m/z) | Eff.b (%) | Other ions (m/z) |
|---|---|---|---|---|---|
| a Vapor pressure values from Syracuse Physical Properties Database (PHYSPROP).26 b Efficiency values (kexp/kcoll), as reported by Thölmann and Grützmacher,18 where kcoll is the collision rate constant. | |||||
| Bromobenzene | 5.57 | C6H5Br +˙ (156) | C6H5NH3+ (94) | 13 | C6H5+ (77) |
| Bromobenzene-d5 | N/A | C6D5Br +˙ (161) | C6D5NH3+ (99) | N/A | C6D5+ (82) |
| Chlorobenzene | 33.3 | C6H5Cl +˙ (112) | C6H5NH3+ (94) | 13 | C6H5+ (77) |
| 1,2-Dichlorobeneze | 1.18 | C6H4Cl2+˙ (146) | C6H4ClNH3+ (128) | 15 | C6H4Cl+ (111) |
| 1,2,4-Trichlorobenzene | 0.613 | C6H3Cl3+˙ (180) | C6H3Cl2NH3+ (162) | N/A | C6H3Cl2+ (145) |
Reaction of 1,2-dichlorobenzene with ammonia
Of the four non-deuterated halobenzenes, 1,2-dichlorobenzene showed the most promise as a reagent for the detection and quantification of ammonia in the gas phase for several reasons. First, the product ions for this reaction were readily detected by the instrument over a range of analyte concentrations (0 to 503 ppm) using product ion accumulation time (PIAT) values 0 to 1000 ms. In addition, both the reagent ion (monoisotopic peak, m/z 146, C6H4Cl2+˙) and the product ion (m/z 128, C6H4ClNH3+) had masses in the desired range, being well over m/z 50. Both the molecular and product ion have characteristic chlorine isotopic signature peaks, which is useful for increased assurance of identification. A single stage mass spectrum for 1,2-dichlorobenzene along with spectra showing the products of reaction of 1,2-dichlorobenzene molecular ion with ammonia at two different concentrations (14 and 503 ppm) is shown in Fig. 2.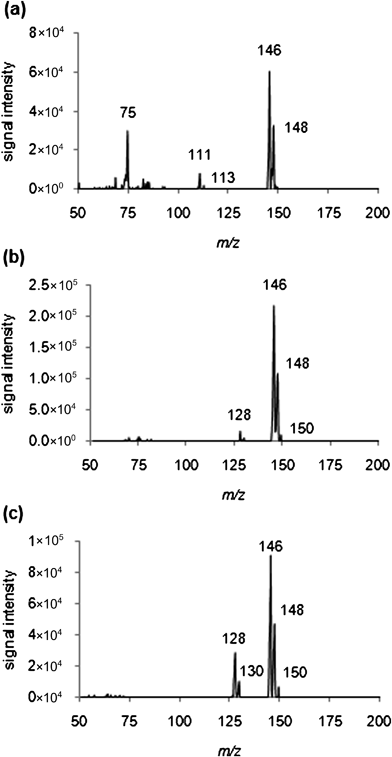 | ||
| Fig. 2 (a) Single stage scan mass spectrum of 1,2-dichlorobenzene (monoisotopic peak, m/z 146, C6H4Cl2+˙) where the LMCO was m/z 50 and ions up to m/z 300 were mass analyzed. Fragment ions at m/z 75 (C6H3+) and 111 (C6H4Cl+), as well as molecular ion at m/z 146 and 148 (isotope signature of two chlorine ions), are present. Mass spectra of 1,2-dichlorobenzene reacting with ammonia at product ion accumulation time of 1000 ms, at ammonia concentrations of (b) 14 ppm and (c) 503 ppm. Present are the reagent molecular ion peaks (at m/z 146, 148, 150) and the product ion peak (m/z 128, C6H4ClNH3+) where chlorine isotopic signatures are evident (direct leak, Griffin 300). Isotope ratios may be somewhat skewed from that predicted, perhaps due to space charge effects at high ion levels. Compare to Fig. S9†, acquired at lower signal levels, m/z 146/148/150, which shows the expected isotope signature (9:6:1) for a dichloro species. | ||
Tandem mass spectrometry
MS2 analysis was used to characterize the product monoisotopic ion (C6H435ClNH3+) at m/z 128 and its corresponding isotope (m/z130,C6H437ClNH3+). The product ion spectrum of the isolated precursor ion (Fig. 3) showed two peaks of interest. The first was the fragment ion (C6H4NH3+˙) from the loss of a chlorine atom from the benzene ring at m/z 93. The second ion at m/z 130 corresponds to the residual chlorine isotope (C6H437ClNH3+) signal in the isolated product ion.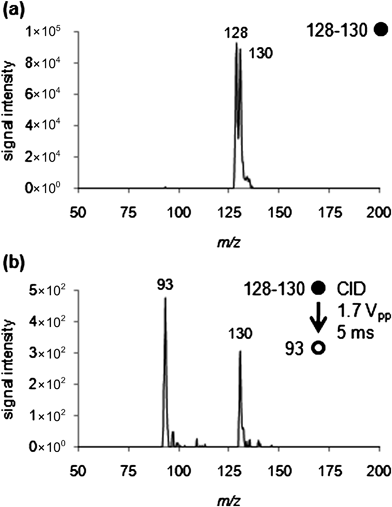 | ||
| Fig. 3
MS2 analysis of 1,2-dichlorobenzene reacting with headspace vapor above aqueous ammonia solution (vapor pressure27 at 21 °C, 743 mbar; ∼700000 ppm). (a) Isolation of m/z 128 and its corresponding isotope, m/z 130, corresponding to the product ion (C6H4ClNH3+). (b) Product ion spectrum of the isolated precursor ion (m/z 128). The fragment ion (C6H4NH3+˙) peak at m/z 93 corresponds to loss of a chlorine atom from the benzene ring. A peak is also seen at m/z 130, which is from the chlorine isotope (C6H437ClNH3+) of the isolated product ion (direct leak, Griffin 300). | ||
Instrument and method response toward ammonia
It was not initially known if ammonia could be pre-concentrated on the Tenax TA/Carboxen 569 sorbent tube bedding, although it was a reasonable expectation that it would. A sorbent tube method typically used on the ChemSense 600 was used to verify that ammonia did adsorb in the tube and the product ion for the reaction with 1,2-dichlorobenzene was observed (Fig. 4). The ion chronogram shows that the product ion abundance increased while the reagent ion abundance simultaneously decreased from about 1.15 to 1.3 min (Fig. 4a), which corresponds to the time period during which the sorbent material was heated. The data appear to indicate that reagent ions were being depleted and a reaction with ammonia was occurring as the ammonia was desorbed from the sorbent material. At about 1.3 min, the reagent ion abundance recovers and the product ion abundance simultaneously decreases, indicating completion of the reaction and suggesting that desorption of the neutral ammonia had been completed. Fig. 4b illustrates a spectrum for the reaction of 1,2-dichlorobenzene with ammonia (5 ppm) at 1.2 min on the ion chronogram.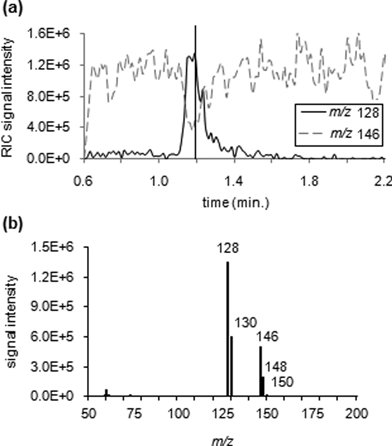 | ||
| Fig. 4 Verification of adsorption of ammonia onto the Tenax TA/Carboxen 569 in the sorbent tube and subsequent desorption. (a) Reconstructed ion chronograms showing reaction of 1,2-dichlorobenzene with ammonia (5 ppm): product ion (m/z 128, dark solid line) and reagent ion (m/z 146, light dashed line). (b) Mass spectrum showing signal for both the reagent cation (m/z 146, 148, and 150) and the reaction product (m/z 128 and 130) at 1.2 min (indicated by the vertical line in Fig. 4a). Method settings: product ion accumulation time 200 ms, ionization time max 150 ms as controlled by the software, 30 s sampling time, 20 s desorption time, and 150 °C desorption temperature (ChemSense 600). | ||
After verifying the reaction, the sorbent tube method was investigated further and optimized. An investigation of the ammonia sampling time over a period of a few seconds to 4 minutes showed that the maximum product ion signal was observed within 90 s of sampling, as indicated by the ammonia reaching the breakthrough volume of the sorbent tube (i.e., the volume sampled beyond which ammonia collection was no longer quantitative). However, it was decided that a sampling time of 30 s, where product ion signal was 75% of the maximum, would be more practical for future applications in which a total method time of about one minute would be desirable. A carryover study showed that the product ion signal dropped below 10% for the second air blank ran after an experiment in which product ion was formed. It was also discovered that allowing the computer's ALC algorithm to control the ionization time vs. a fixed time (e.g., 1.5 ms) was ideal. A PIAT value of 200 ms, desorption time of 20 s, and desorption temperature of 150 °C proved to give good overall product ion signal intensity. This optimized method was then used to quantify the instrument's response toward ammonia.
Linear calibration curve fit parameters and other useful quantitative information are recorded in Table 2 for a direct leak and sorbent tube method. Parameters include: limits of linearity (LOL; i.e., the upper concentration limit of the linear response region),28 LOD25 at the 95% confidence level, average of the blank (aveblank), standard deviation of the blank (σblank), the number of data points recorded (N), the slope of the fit line (m), standard deviation of the slope (σm), intercept of the fit line (b), standard deviation of the intercept (σb), and the correlation coefficient (R2). The linear dynamic range (LDR) for an analyte may be obtained by subtracting the LOD from the LOL. The graphical calibration plots for both the direct leak and sorbent tube method on the ChemSense 600 are shown in Fig. 5.
| Experiment type | LOL d (ppm) | LODe (ppm) | aveblank | σ blank | Calibration curve parametersf | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | m | σ m | b | σ b | R 2 | |||||
| a Data collected for the product ion (m/z 128, C6H4ClNH3+) from the reaction of 1,2-dichlorobenzene with ammonia in all cases. b Direct leak experimental parameters: ionization time fixed at 1.5 ms and product ion accumulation time (PIAT) 200 ms. c Sorbent tube experimental parameters: ionization time controlled by ALC algorithm (maximized at 150 ms), sampling time 30 s, desorption time 20 s at 150 °C, and PIAT 200 ms. d Limit of linearity (LOL),28 where *indicates LOL of the highest concentration tested and **indicates LOL of the highest concentration tested before linearity of the regression fit is visibly lost. e Limit of detection (LOD)25 at the 95% confidence level. f Calibration curve parameters obtained from a linear least squares fit: N = number of data points recorded, m = slope, σm = std. dev. of slope, b = intercept, σb = std. dev. of intercept, and R2 = correlation coefficient. | ||||||||||
| Direct leakb | 503 | 17 | 2.0 × 101 | 3.3 × 101 | 1028 | 6.0 × 100 | 4.4 × 10−2 | 2.6 × 101 | 1.0 × 101 | 0.9483 |
| Sorbent tubec | 10* | 0.54 | 1.8 × 104 | 1.2 × 104 | 38 | 2.2 × 106 | 1.0 × 105 | 7.8 × 105 | 3.6 × 105 | 0.9281 |
| Sorbent tubec | 5** | 0.22 | 1.8 × 104 | 1.2 × 104 | 35 | 3.0 × 106 | 9.7 × 104 | 3.2 × 104 | 2.0 × 105 | 0.9677 |
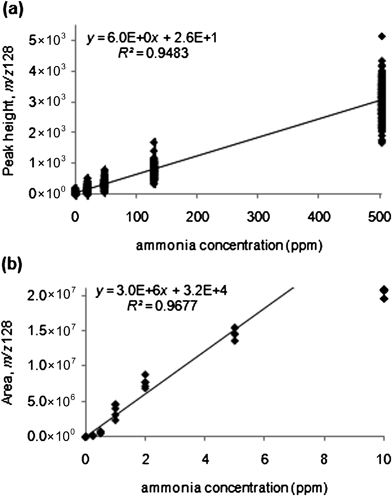 | ||
| Fig. 5 Calibration curves for the reaction of 1,2-dichlorobenzne with ammonia. Product ion signal (m/z 128, C6H4ClNH3+) plotted vs. concentration of ammonia for (a) direct leak (LOD 17 ppm). Direct leak experimental parameters: ionization time fixed at 1.5 ms and product ion accumulation time (PIAT) 200 ms. (b) Sorbent tube (LOD 220 ppb; fit to 5 ppm) experiment. Sorbent tube experimental parameters: ionization time controlled by ALC algorithm (maximized at 150 ms), sampling time 30 s, desorption time 20 s at 150 °C, and PIAT 200 ms (ChemSense 600). | ||
The LOD for the direct air leak was 17 ppm with 95% confidence on the ChemSense 600 (comparable to the 30 ppm LOD obtained on the Griffin 300, data not shown). However, the pre-concentration sorbent tube method of the ChemSense 600 was found to be much more sensitive, as expected, with a LOD of 540 ppb for a linear fit up to the highest ammonia concentration investigated (10 ppm). Linearity, however, was clearly lost between 5 and 10 ppm (Fig. 5b). A calibration fit up to 5 ppm yielded a LOD of 220 ppb with 95% confidence, which was just under half of that for the fit up to 10 ppm (Table 2). Leveling of the calibration curve at larger concentrations suggests that the reagent ion is being exhausted around 5 ppm. Overall, these LODs compare well with the IDLH (300 ppm), PEL (50 ppm), and odor threshold (5 ppm) levels of ammonia.6
Receiver operating characteristic curves
Receiver operating characteristic curves were originally developed to better distinguish amongst noisy radio signals29 and later used in medical settings.30,31 More recently, a modified type of ROC curve, as applied to chemical and biological sensors, was proposed by the U.S. Department of Defense (DoD),32 which we refer to as “DoD–ROC curves”. These curves may be used to assess and compare instrument performance. The curves plot analyte concentration versus false positive probability for a given detection likelihood. The method of computing these curves is described in more detail elsewhere.33,34 If a mass spectrometer,9,24,33 for example, has high sensitivity and thus a low false positive probability as concentrations approach that of the blank, the DoD–ROC curve will have an “L” shape in the upper left hand corner of the plot (i.e., the plot will hug the top and left axes). By presenting and comparing such DOD–ROC curves between two hypothetical methods or instruments, confusion resulting from different definitions of the LOD (still common where practitioners from different fields are involved) should be eliminated. Two such plots with 95% detection likelihood are shown in Fig. 6 where the concentration of ammonia is plotted vs. false positive rate for the reaction of 1,2-dichlorobenzene with ammonia for both a direct leak and sorbent tube experiment. Note the different vertical scales.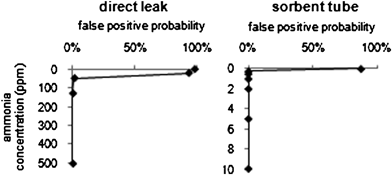 | ||
| Fig. 6 DoD–ROC curves for the reaction of 1,2-dichlorobenzne with ammonia for a direct leak (left) and sorbent tube experiment (right). Concentration of ammonia is plotted versus false positive probability for each concentration at 95% detection likelihood (ChemSense 600). Note the different vertical scales. | ||
Assessment of competing reaction paths
It was of interest to ascertain the contribution of fragment ions (viz., the single-chlorine loss ion at m/z 111, C6H4Cl+) to the formation of reaction product at m/z 128. This was probed by changing the low mass cutoff (LMCO) during the product ion accumulation time (PIAT) in the reaction scan function (Fig. 1, segment 5). For the control experiment, the low mass cutoff of the PIAT segment was held at m/z 50 as usual. A second experiment was conducted in which the PIAT LMCO was set to m/z 125, which was just below that of the product ion (m/z 128, C6H4ClNH3+) and above the m/z ratios of all other fragment ions in the single-stage mass spectrum (Fig. 3a). Raising the LMCO will eject ions at m/z 111 (C6H4Cl+) from the CIT, preventing the creation of product ions from the combination of m/z 111 with ammonia.For each experiment, product ion signal, normalized with respect to total ion signal, was plotted as a function of PIAT. We expected the normalized product ion signal to increase as time elapsed during the PIAT, since the reaction would have a longer time to occur. If, on the other hand, the increase of product ion in the two experiments were the same, this would suggest that the fragment ion (m/z 111) was not contributing to product formation, with the product ion (m/z 128) being formed solely from the parent ion (m/z 146, C6H4Cl2+˙) reacting with ammonia. Conversely, a horizontal line (slope = 0) when the PIAT LMCO was held at m/z 125 would indicate the product ion was formed solely by the m/z 111 fragment ion, with m/z 146 playing no role in product formation.
When the PIAT LMCO was held at m/z 125, normalized product ion signal vs. PIAT rose somewhat less rapidly than the normalized product signal vs. PIAT with LMCO = m/z 50 (Fig. S7†). Thus, it may be concluded that by 600 ms product ion accumulation time, roughly two-thirds of the product ion has been formed by the reaction of m/z 146 with ammonia and roughly one-third by the reaction of m/z 111 with ammonia. As all fragment ions are swept from the CIT during segment 4 of the scan function, we surmise that ions of m/z 111 must be formed by the dissociation of m/z 146 during the PIAT. We also observe that the normalized parent ion abundance decreases at about 0.6 times the rate by 600 ms PIAT when the LMCO was held at m/z 125. When the LMCO is raised to m/z 125, the potential well depth seen by parent ions (m/z 146, C6H4Cl2+˙) is much greater than that seen by those same ions when the LMCO was only m/z 50. Thus, it is likely that the ions at m/z 146 were trapped more efficiently, resulting in higher signal for m/z 146 when the PIAT LMCO was set to m/z 125.
Reaction of 1,2-dichlorobenzene with ammonia and two amines
1,2-Dichlorobenzene was reacted with ammonia, methylamine, and ethylamine (all at 503 ppm) in the gas phase as described in the Experimental Section. The expected product ions formed for methylamine (m/z 142, C6H4ClNH2CH3+) and ethylamine (m/z 156, C6H4ClNH2CH2CH3+) presumably follow a reaction analogous to reaction (1) (Fig. S8†). The product ion abundances for methylamine and ethylamine increased much more quickly with time as compared to that for ammonia. Overall, the ethylamine and methylamine products are favored almost equally but more so than the ammonia product when all three species are mixed together.Nitrile reactions
We investigated two reactions with nitriles analogous to that presented in reaction (1).000 ppm) can undergo an analogous substitution reaction with the 1,2-dichlorobenzene cation (reaction (2), Fig. S9†). The product ion at m/z 152 corresponded to [C6H4ClNCCH3]+ and an isotopic signature peak for chlorine was also seen at m/z 154. However, the signal intensity was very low for m/z 154, which meant using this chlorine isotope peak as a confirmation peak of the product would most likely not be possible.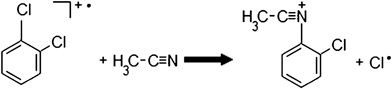 | (2) |
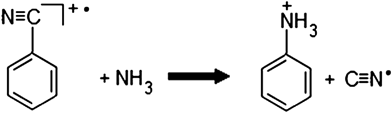 | (3) |
However, no product ion (m/z 94 [C6H5NH3]+) was seen for the reaction of 503 ppm ammonia with headspace benzonitrile (C6H5CN; FW 103 g mol−1; vapor pressure26 at 25 °C, 1.3 mbar; ∼1300 ppm). Product ion signal, however, was present for the reaction of headspace ammonia (∼700000 ppm) with headspace benzonitrile (Fig. S10†).
These results indicate that ion/molecule reactions similar to those presented in reaction (1) are potentially appropriate for nitriles along with other pseudohalogenides. A more reactive pseudohalogenide would, perhaps, have better results than those seen with benzonitrile. However, a reaction comparable to reaction (2) shows good promise for the detection of acrylonitrile (CH2CHCN, FW 53 g mol−1), which is also a DHS toxic industrial compound.
Conclusions
Two miniature mass spectrometers (Griffin 300 and ChemSense 600) equipped with internal ionization sources (filament EI and GDEI) and CIT mass analyzers were successfully used to detect, indentify, and quantify gaseous, neutral ammonia. This was accomplished by way of an ion/molecule reaction with halobenzene radical cations (bromobenzene, bromobenzene-d5, chlorobenzene, 1,2-dichlorobenzene, and 1,2,4-trichlorobenzene) to give a product that was readily detected relative to the instrument's low mass cutoff of ∼m/z 40. LODs were below IDLH and PEL of ammonia for direct air leak (17 ppm) and below the odor threshold for sorbent tube pre-concentration (220 ppb). To our knowledge, this is the first time this type of method has been used to detect ammonia. Instrument performance was also described using DOD–ROC curves.Implementing this method on a mass spectrometer that is fully automated and fieldable, such as the ChemSense 600, should prove useful for applications such as facility air monitoring where the detection of toxic compounds is of particular interest. Though effective, the need for the lengthy customized reaction scan function (total length up to 1240 ms) may limit this method's applicability with the current ChemSense 600 method, whereby desorption from a thermal sorption/desorption trap occurs over 15 seconds and is alternated with a 5 second direct air leak sampling step. One possible implementation would be to trigger the special scan function to run after the identification of ammoniavia an alternative method, e.g. an electrochemical sensor. Obviously, this method also requires an on-board vial of a halobenzene, which could be implemented on the ChemSense 600. The current PFTBA mass calibrant compound vial could be replaced with a vial containing the liquid halobenzene reagent. The idea of using an ion/molecule reaction to increase the effective m/z of the species to be detected may also be applicable to other low-mass TICs [e.g., formaldehyde (CH2O, FW 30 g mol−1), hydrogen cyanide (HCN, FW 27 g mol−1), and acrylonitrile (CH2CHCN, FW 53 g mol−1)]. Modification of the instrument to allow for negative ion detection would also open up the possibility for other types of reaction products to be detected.
Acknowledgements
Funding for this project was provided by the Department of Homeland Security/Science and Technology Division (HSHQPA-05-9-0033) and by ICx Analytical Instruments. The authors also express their gratitude to Jason Duncan (Purdue University) for engineering support with the Griffin 300 and ICx for use of the ChemSense 600.References
- Z. Ouyang and R. G. Cooks, Annu. Rev. Anal. Chem., 2009, 2, 187–214 Search PubMed.
- R. E. March and J. F. Todd, Quadrupole Ion Trap Mass Spectrometry, John Wiley and Sons, Inc., New Jersey, 2nd edn, 2005 Search PubMed.
- H. W. Chen, R. F. Xu, H. Chen, R. G. Cooks and Z. Ouyang, J. Mass Spectrom., 2005, 40, 1403–1411 CrossRef CAS.
- L. Gao, Q. Y. Song, G. E. Patterson, R. G. Cooks and Z. Ouyang, Anal. Chem., 2006, 78, 5994–6002 CrossRef CAS.
- L. S. Riter, E. C. Meurer, E. S. Handberg, B. C. Laughlin, H. Chen, G. E. Patterson, M. N. Eberlin and R. G. Cooks, Analyst, 2003, 128, 1112–1118 RSC.
- NIOSH, NIOSH Pocket Guide to Chemical Hazards, Publication No. 97–140, National Institute for Occupational Safety and Health, Cincinnati, OH, 2005, http://www.cdc.gov/niosh/npg/, accessed July, 2010 Search PubMed.
- M. Appl, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
- Nist, Nist Chemistry WebBook, http://webbook.nist.gov/chemistry/, accessed July, 2010.
- I. Cotte-Rodríguez, D. R. Justes, S. C. Nanita, R. J. Noll, C. C. Mulligan, N. L. Sanders and R. G. Cooks, Analyst, 2006, 131, 579–589 RSC.
- H. F. Grutzmacher and M. Buchner, Eur. J. Mass Spectrom., 2004, 10, 21–26 CrossRef.
- H. F. Grutzmacher, Curr. Org. Chem., 2003, 7, 1565–1587.
- A. Nixdorf and H. F. Grutzmacher, Int. J. Mass Spectrom., 2000, 195, 533–544 Search PubMed.
- M. Buchner, A. Nixdorf and H. F. Grutzmacher, Chem.–Eur. J., 1998, 4, 1799–1809 CrossRef CAS.
- L. Bache-Andreassen and E. Uggerud, Int. J. Mass Spectrom., 2000, 195, 171–184 Search PubMed.
- T. Drewello, N. Heinrich, W. P. M. Maas, N. M. M. Nibbering, T. Weiske and H. Schwarz, J. Am. Chem. Soc., 1987, 109, 4810–4818 CrossRef CAS.
- R. Lopez, E. del Rio, M. I. Menendez and T. L. Sordo, J. Phys. Chem. A, 2002, 106, 4616–4622 CrossRef CAS.
- G. P. Slater and J. F. Manville, J. Chromatogr., A, 1993, 648, 433–443 CrossRef CAS.
- D. Thölmann and H. F. Grützmacher, J. Am. Chem. Soc., 1991, 113, 3281–3287 CrossRef.
- G. E. Patterson, A. J. Guymon, L. S. Riter, M. Everly, J. Griep-Raming, B. C. Laughlin, Z. Ouyang and R. G. Cooks, Anal. Chem., 2002, 74, 6145–6153 CrossRef CAS.
- L. S. Riter, Y. Peng, R. J. Noll, G. E. Patterson, T. Aggerholm and R. G. Cooks, Anal. Chem., 2002, 74, 6154–6162 CrossRef CAS.
- G. E. Patterson, J. M. Wells, J. W. Grossenbacher, A. J. Cochran, B. A. Knecht, B. D. Rardin, J. S. Springston and D. J. Barket, Conference on Detector/Sensor Research and Technology for Homeland and National Security, Gatlinburg, TN, September 14–16, 2004.
- L. Gao, Q. Y. Song, R. J. Noll, J. Duncan, R. G. Cooks and O. Y. Zheng, J. Mass Spectrom., 2007, 42, 675–680 CrossRef CAS.
- L. Gao, A. Sugiarto, J. D. Harper, R. G. Cooks and Z. Ouyang, Anal. Chem., 2008, 80, 7198–7205 CrossRef CAS.
- J. N. Smith, A. Keil, J. Likens, R. J. Noll and R. G. Cooks, Analyst, 2010, 135, 994–1003 RSC.
- L. A. Currie, Anal. Chem., 1968, 40, 586–593 CrossRef CAS.
- PHYSPROP, Syracuse Physical Properties Database, http://www.srcinc.com/what-we-do/product.aspx%3Fid%20%3D%202133, accessed January, 2010.
- BBC Biochemical, MSDS Report for Ammonium Hydroxide, http://www.bbcus.com/uploads/files/AmmoniumHydroxide_MSDS.pdf, accessed December, 2009.
- D. A. Skoog, F. J. Holler and T. A. Nieman, Principles of Instrumental Analysis, Harcourt Brace & Company, Philadelphia, PA, 5th edn, 1998 Search PubMed.
- J.v. Schalkwyk, The Magnificent ROC, 2005, http://www.anaesthetist.com/mnm/stats/roc/Findex.htm, accessed August, 2009 Search PubMed.
- S. C. Kazmierczak and P. G. Catrou, Anal. Chem., 1995, 67, R437–R441.
- N. A. Obuchowski, M. L. Lieber and F. H. Wians, Clin. Chem. (Washington, DC, U. S.), 2004, 50, 1118–1125 CrossRef CAS.
- J. Carrano, Chemical and Biological Sensor Standards Study, DARPA Microsystems Technology Office, U.S. Deparment of Defense, 2004, http://www.darpa.mil/mto/publications/ Search PubMed.
- C. C. Mulligan, D. R. Justes, R. J. Noll, N. L. Sanders, B. C. Laughlin and R. G. Cooks, Analyst, 2006, 131, 556–567 RSC.
- I. Cotte-Rodríguez, D. R. Justes, S. C. Nanita, R. J. Noll, C. C. Mulligan, N. L. Sanders and R. G. Cooks, Analyst, 2006, 131, 579–589 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: A more detailed explanation of the sample preparation and introduction system subsection; single stage and reaction mass spectra for bromobenzene, bromobenzene-d5, chlorobenzene, and 1,2,4-trichlorobenzene, and spectra for the two nitrile reactions that were explored. See DOI: 10.1039/c0an00630k |
| This journal is © The Royal Society of Chemistry 2011 |