Towards the electrochemical quantification of the strength of garlic
Benjamin C. M.
Martindale
,
Leigh
Aldous
,
Neil V.
Rees
and
Richard G.
Compton
*
Department of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: richard.compton@chem.ox.ac.uk; Fax: +44 (0)1865 275 410; Tel: +44 (0)1865 275 413
First published on 3rd November 2010
Abstract
A simple but sensitive technique has been demonstrated towards the electroanalytical quantification of the strength of garlic. This technique can also be used to quantify dialkyldisulfides. The cyclic voltammetry of bromide was found to be a sensitive electrochemical probe, electrogenerated bromine reacting with dialkyldisulfides to catalytically regenerate bromide, resulting in a significant increase in peak current. A linear response of current vs. concentration was observed between 0.1 and 15 mM dipropyldisulfide at edge plane pyrolytic graphite (EPPG) electrodes; a smaller range up to ca. 5 mM was available at screen printed carbon electrodes (SPCEs), with a detection limit (from 3σ) of 0.067 mM. The response of diallyldisulfide was found to be essentially identical. Shaking garlic puree in acetonitrile for 5 minutes, followed by dilution with water then recording the voltammetry at the cheap, disposable SPCE gave a linear trend in current with respect to the quantity of garlic present, corresponding to the diallyldisulfide extracted. This has potential applications in monitoring the garlic content of medicinal supplements, batch-to-batch variation and the stability of garlic during storage.
Introduction
Garlic is consumed in a variety of forms for its medicinal properties, and is also used worldwide as a cooking ingredient.1–3 Data indicates that in 2007 at least 15.7 million tonnes of garlic was produced worldwide.2 However, different batches of garlic vary significantly in the strength of flavour, therefore the ‘garlic flavour’ needs to be quantified before appropriate amounts of garlic can be added during industrial-scale food preparation. This fact, as well as variations caused by different cooking methods3 and the physiological and biochemical activity of the various components of garlic,2,3 has led to interest in a variety of fields in the quantification of the characteristic flavour and fragrance molecules of garlic.In the food industry garlic batches are frequently investigated by organoleptic testing using a trained panel of human experts. One method is based upon the dilution of garlic in sour cream followed by taste testing; repeated testing can be used in order to monitor the stability of garlic during long-term storage. The extraction, identification and quantification of the volatile flavour constituents of garlic have been described by a number of authors,2 for example by solid-phase trapping solvent extraction,1headspace solid-phase microextraction,1steam distillation,1steam distillation/solvent extraction1,3 and super-critical CO2 extraction.4,5 It has been found that the majority of these volatile compounds are thiosulfinates, although the rich variety of compounds that contribute to the flavour and fragrance of garlic stem from the odourless precursor alliin. Upon rupture of the garlic cellular tissue alliin is converted enzymatically to allicin,1 which makes up ca. 70% of the thiosulfinates present in freshly chopped garlic (ca. 0.4 to 1.4% w/w of the garlic itself).2 However, allicin is also unstable and decomposes to form a variety of compounds; although the quantitative determination of allicin has been described,4 most studies determine diallyldisulfide to be the most overwhelmingly abundant extractant from processed, raw garlic,1,3,5 as well as garlic that has been cooked by a number of methods.3,5
The heat of various chilli sauces was recently quantified by the adsorptive stripping voltammetry (ASV) of the key ‘hot’ ingredient, capsaicin.6 An equivalent technique to investigate garlic would require extraction and quantification of the predominate sulfur-containing species found in chopped or processed garlic, namely diallyldisulfide.
A number of techniques have been described for the electrochemical quantification of disulfides.7Disulfides such as cystine8,9 and oxidised glutathione8,10 have been quantified by their accumulation then stripping at hanging mercury drop electrodes. Pulsed electrochemical detection has also been used for the quantification of cystine and oxidised glutathione at gold electrodes.11–14 However, such techniques utilise toxic or expensive metals, and are also likely to be susceptible to interference by the non-thiosulfinate constituents of garlic.
Bromine has been electrochemically generated, reacted on column and the remaining bromine detected downstream, in order to indirectly quantify thiols (cysteine and reduced glutathione) and the dialkylmonosulfide methionine.15Iodine was used in a similar manner to quantify cysteine and N-acetylcysteine in urine.16Bromine has also been used as a mediator during electrosynthesis; cysteic acid was prepared by the reaction of electrogenerated bromine with cystine,17 while reaction with a range of substituted diaryldisulfides was reported to form sulfenium cations as intermediates.18 The reaction of molecular bromine with disulfides has also been used for the direct titration (as thus quantification) of solutions of dialkyldisulfides.19 The authors are unaware of any published work relating to the amperometric quantification of disulfidesvia the electrogeneration of bromine.
In this work we have investigated the quantification of two disulfides (diallyldisulfide and dipropyldisulfide) using the electrochemistry of sodium bromide as a mediator. Bromine is generated in situ at carbon electrodes, hence there are no health concerns compared to the use of molecular bromine or mercury electrodes. Investigation of the system has demonstrated that following electron transfer to form bromine, the disulfides react with several equivalents of bromine to regenerate bromide, which is then reoxidised at the electrode (similar to an EC mechanism20). Proof-of-principle in terms of quantifying the strength of garlic has also been demonstrated; shaking a sample of raw garlic puree in acetonitrile for 5 min followed by voltammetric investigation at cheap, disposable screen-printed carbon electrodes was linear with respect to the quantity of garlic present, and will presumably relate to the strength of the garlic (e.g. in terms of w/w% in certain dishes, stability during long-term storage, or batch-to-batch and seasonal variations).
Experimental
All chemicals were purchased at the highest grade available and used directly without any further purification. Dipropyldisulfide (98%), diallyldisulfide (tech., 80%) and NaClO4 (ACS Reagent, ≥98%) were sourced from Sigma Aldrich, UK; NaBr was sourced from May and Baker Ltd, UK. All solutions were prepared with acetonitrile (HPLC Gradient grade, Fisher Scientific) and deionised water of resistivity not less than 18.2 MΩ cm−1 at 298 K (Millipore UHQ, Vivendi, UK).Garlic samples were kindly donated by Beacon Foods Ltd (Brecon Powys, UK), and consisted of two batches of garlic sourced from China and Spain. The garlic cloves were peeled and chopped in Beacon Foods Ltd kitchens then posted under refrigerated conditions. The garlic was stored at 4 °C, and all experiments were performed within five days of receipt. Extraction was performed as described in the Results and discussion section.
All voltammetric measurements were recorded using a µ-Autolab III computer-controlled potentiostat (EcoChemie, Utrecht, The Netherlands). The DRP-110 Screen Printed Carbon Electrodes (SPCEs) were manufactured by DropSens Ltd (Spain) and obtained through their UK distributor (IJ Cambria Scientific Ltd, UK). The edge plane pyrolytic graphite (EPPG, Le Carbone Ltd., Sussex, UK) and basal plane pyrolytic graphite (BPPG, Le Carbone Ltd., Sussex, UK) electrodes were prepared by securing the material inside an insulating PTFE surround to leave an exposed circular geometric surface with a diameter of 4 mm (EPPG) or 5 mm (BPPG). The EPPG electrode surface was renewed by polishing with alumina slurries (1.0–0.3 µm, Buehler Ltd., USA). The BPPG electrode surface was renewed by firmly pressing the surface on cellotape then pulling off to remove the surface layers, which was repeated five times before thoroughly rinsing the surface with acetone and then deionised water.
Experiments were performed using a three-electrode set-up, with a graphite rod and leak-free Ag/AgCl (Cypress Systems, USA) as counter and reference, respectively. A beaker containing 16 mL or 25 mL of electrolyte was employed at ambient temperature; at no stage was degassing found to be necessary.
Results and discussion
Comparison of the oxidation of bromide at edge plane pyrolytic graphite (EPPG), basal plane pyrolytic graphite (BPPG) and dropSens screen printed carbon electrodes (SPCEs)
The ultimate goal was to prepare a garlic sensor using screen printed carbon electrodes (SPCEs). SPCEs are typically cheap, customisable and disposable.21,22 Having the necessary three electrodes printed onto a small, easy-to-connect strip, makes them easy to handle and use for food quality control procedures.23,24 However, in order to obtain additional information regarding the nature of the system, the employed DropSens SPCEs were also compared against edge plane pyrolytic graphite (EPPG) and basal plane pyrolytic graphite (BPPG) electrodes. Fig. 1 displays an overlay of CVs recorded using the three carbon electrode materials for the oxidation of Br− in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water:acetonitrile (H2O:AcN). Oxidation of Br− at BPPG occurred as a well-defined peak at ca. +1.33 V, while oxidation at the EPPG and SPCE both gave rise to two overlapping features, at ca. +0.95 and ca. +1.15 V. The first oxidative feature at the EPPG and SPCE is attributed to the oxidation of Br− to Br2, followed by rapid reaction with Br− to form [Br3]−, while the second feature corresponds to the oxidation of [Br3]− to yield Br2. Such behaviour is well established in acetonitrile solutions.25,26 The similarity of the EPPG and SPCE responses also agrees with the previous findings by Fanjul-Bolado et al.27 and Kadara et al.,28 namely that DropSens SPCEs have a large area of edge planes exposed to solution due to the high density and likely random orientation of the graphite platelets of which the electrode consists.
:50 water:acetonitrile (H2O:AcN). Oxidation of Br− at BPPG occurred as a well-defined peak at ca. +1.33 V, while oxidation at the EPPG and SPCE both gave rise to two overlapping features, at ca. +0.95 and ca. +1.15 V. The first oxidative feature at the EPPG and SPCE is attributed to the oxidation of Br− to Br2, followed by rapid reaction with Br− to form [Br3]−, while the second feature corresponds to the oxidation of [Br3]− to yield Br2. Such behaviour is well established in acetonitrile solutions.25,26 The similarity of the EPPG and SPCE responses also agrees with the previous findings by Fanjul-Bolado et al.27 and Kadara et al.,28 namely that DropSens SPCEs have a large area of edge planes exposed to solution due to the high density and likely random orientation of the graphite platelets of which the electrode consists.
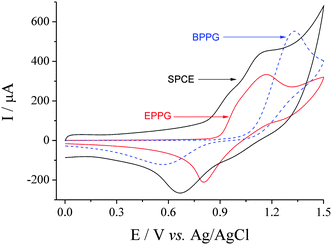 | ||
| Fig. 1
CVs of 15 mM NaBr in 50:50 H2O:AcN (0.1 M NaClO4) at SPCE (diameter 4 mm), EPPG (4 mm) and BPPG (5 mm) electrodes. Recorded at 50 mV s−1. | ||
Mediated oxidation of dipropyldisulfide (PrSSPr) with bromine at SPCE: concentration and square wave voltammetric analysis
Fig. 2 displays CVs recorded for 15 mM Br− in 50:50 H2O:AcN with the standard addition of dipropyldisulfide (PrSSPr), using a new SPCE with each addition. The largest oxidative peak (corresponding to Br2 generation) increased in a linear manner with the amount of PrSSPr added, while the reverse peak (reduction of Br2) decreased. The oxidation peak grew and shifted more positive with each addition of PrSSPr, with the oxidation peak eventually becoming indistinguishable from the solvent oxidation at ca. 5 mM PrSSPr (only shown up to 2 mM PrSSPr in Fig. 2). The PrSSPr displayed no voltammetric features of its own on the SPCE in the investigated potential range. Such change in the behaviour of Br− is qualitatively indicative of an electron transfer to form Br2, followed by chemical reaction to catalytically regenerate the initial species (either Br− or [Br3]−), abbreviated as an EC reaction20 and discussed in more detail later.
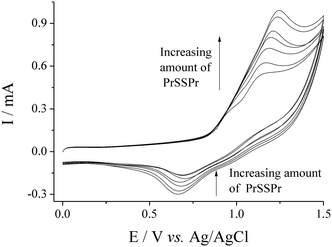 | ||
| Fig. 2
CVs for the oxidation of 15 mM NaBr in 50:50 H2O:AcN (0.1 M NaClO4) at SPCE with the standard addition of dipropyldisulfide (0, 0.33, 0.66, 1.00, 1.33, 1.66 and 1.99 mM). Recorded at 50 mV s−1. | ||
Square wave voltammetric (SWV) analysis of the system was also carried out. First the parameters were optimised in order to give the largest current response and most clearly resolved peaks. During this process it was observed that the oxidation processes were relatively irreversible, such that the initial and subtracted current values were similar and the resulting SWV had a significantly smaller current than the initial current alone. Fig. 3 displays an overlay of the forward current with the resulting SWV for 15 mM Br− in 50:50 H2O:AcN at a SPCE, using the optimised SWV parameters. The oxidation of Br− to [Br3]− (ca. +0.95 V) and Br2 (ca. +1.25 V) are clearly resolved in the SWV. An additional oxidative feature is also observed at ca. +0.75 V. This process cannot be observed in the forward current alone as it is masked by the double layer charging, and likely relates to the adsorption of Br− at the rough carbon surface.
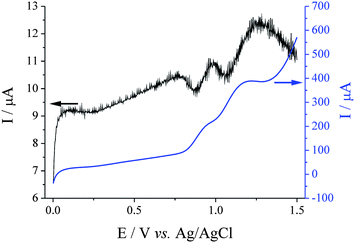 | ||
| Fig. 3 Overlaid square wave scans for 15 mM NaBr in 50:50 H2O:AcN (0.1 M NaClO4) at a SPCE, displaying the forward current only (corresponding to the right-hand y-axis) and the resulting square wave voltammogram. Recorded at a frequency of 25 Hz, an amplitude of 0.9 mV and step potential of 1.95 mV. | ||
SWV were recorded for SPCEs in solutions containing NaBr (15 mM) and PrSSPr (0 to 1.33 mM), using a new SPCE for each solution. It was observed that differences between the individual SPCE, which did not obviously affect the CVs, had significantly more influence on the response recorded using SWV due to the much smaller currents. Most notably the absolute current values were shifted by a different value for each SPCE. Fig. 4(a) displays SWV of Br− in the presence of varying amounts of PrSSPr that have been smoothed and then ‘referenced’, by shifting the entire voltammogram such that the current at a consistent minimum in the SWV at +0.20 V was equal to zero. Fig. 4(b) displays a plot of the peak current, Ipvs.PrSSPr concentration for the peak at ca. +0.95 V (○) and the peak at ca. +1.25 V (■). The results clearly demonstrate that with the addition of PrSSPr the peak corresponding to the formation of [Br3]− remains unchanged while the peak corresponding to Br2 increases in a linear manner, demonstrating that Br2 is the only species that reacts with the PrSSPr. It also highlights that in this case SWV offers no significant advantage over CV in terms of data collection, the increased resolution frequently associated with SWV being counteracted somewhat by batch-to-batch variation in the SPCE.
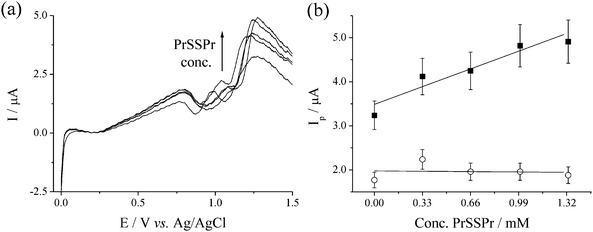 | ||
| Fig. 4 (a) SWV of 15 mM NaBr in 50:50 H2O:AcN (0.1 M NaClO4) at SPCE, in the presence of PrSSPr (0, 0.33, 0.66, 1.00 and 1.33 mM). Scans smoothed and offset such that at E = 0.20 V the current is equal to zero. SWV parameters are the same as in Fig. 3. (b) Plot of Ipvs.PrSSPr concentration for the peaks at ca. +0.95 V (○) and +1.25 V (■, gradient = 1.2 ± 0.2 µA mM−1). Lines are linear best fits, error bars are assuming 10% error. | ||
Comparison of diallyldisulfide with dipropyldisulfide
The primary flavour-component of raw garlic varies based upon the method used to extract and quantify it, although diallyldisulfide (4,5-dithia-1,7-octadiene) consistently ranks as the predominate compound with its extracted abundance between 58 and 98%, in addition to various other thiosulfinates and hydrocarbons.1 Since diallyldisulfide could not be sourced in high purity the structurally similar dipropyldisulfide (98% purity), which is essentially a saturated analogue of diallyldisulfide, was employed for the precise work described throughout the paper. However, examination of the two species was required in order to compare their relative responses, and a technical grade sample of diallyldisulfide with quoted purity of 80% was investigated (primary contaminants anticipated to be the tri- and polysulfide derivatives2).Experiments similar to those described in Fig. 2 were repeated, whereby CVs were recorded for the oxidation of 15 mM NaBr in 50:50 H2O:AcN at SPCE, in the presence of 0 to 2 mM of either dipropyldisulfide or diallyldisulfide, with the diallyldisulfide being treated as 80% diallyldisulfide v/v. Fig. 5(a) displays a plot of peak current for the peak at ca. +1.15 V vs. the concentration of the relevant disulfide species. It can be observed that both species gave closely similar linear responses with respect to concentration of the disulfide compound; both gave identical gradients of 224 µA mM−1, although visually the data points for diallyldisulfide are also consistently lower than those representing equivalent concentrations of dipropyldisulfide. Therefore the two compounds are assumed to have an identical response, with the slight difference in the absolute current–concentration ratio being attributed to the actual diallyldisulfide concentration in the supplied sample being slightly lower than 80% v/v. Different diffusion coefficient values could also account for the variation, although the diffusion coefficients of diallyldisulfide and dipropyldisulfide are not expected to differ significantly.
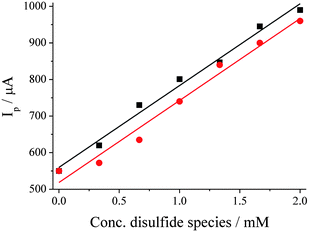 | ||
| Fig. 5 Plot of Ip at ca. +1.15 V vs. concentration of diallyldisulfide (●, gradient = 224 ± 14 µA mM−1, R2 = 0.98) and dipropyldisulfide (■, gradient = 224 ± 10 µA mM−1, R2 = 0.99). CVs recorded in 50:50 H2O:AcN (0.1 M NaClO4) containing 15 mM NaBr at SPCE and 50 mV s−1. | ||
Voltammetry in the presence of 0.005 to 2 mM PrSSPr was investigated at SPCE, which gave the expected gradient of 224 µA mM−1 and an LOD value (based upon 3σ)29 of 0.067 mM.
Confirmation of the EC mechanism by scan rate study and simulation at EPPG
In order to investigate the electrochemical process in more detail, the voltammetry of Br− and PrSSPr was investigated further. An EPPG electrode was used in order to ensure that a well-defined, reproducible electrode surface was used for each scan.With the addition of small quantities of PrSSPr to solutions of Br− a small prepeak was formed. Fig. 6(a) displays CVs of 15 mM NaBr in the presence of 0 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), 0.1, 0.2 and 0.3 mM PrSSPr, in 50:50 H2O:AcN at 50 mV s−1 at EPPG. Fig. 6(b) displays plots of peak current vs.PrSSPr concentration at three different NaBr concentrations, demonstrating that the peak current was linear with PrSSPr concentration except when [PrSSPr] = 0.
), 0.1, 0.2 and 0.3 mM PrSSPr, in 50:50 H2O:AcN at 50 mV s−1 at EPPG. Fig. 6(b) displays plots of peak current vs.PrSSPr concentration at three different NaBr concentrations, demonstrating that the peak current was linear with PrSSPr concentration except when [PrSSPr] = 0.
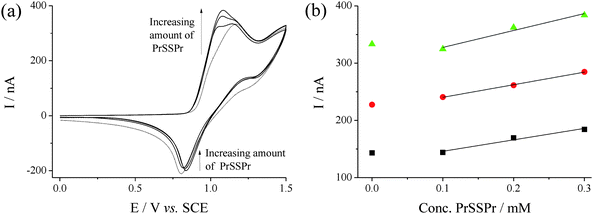 | ||
| Fig. 6 (a) CVs of 15 mM NaBr in 50:50 H2O:AcN (0.1 M NaClO4) at an EPPG electrode, in the presence of PrSSPr (0, 0.10, 0.20 and 0.30 mM), recorded at 50 mV s−1, and (b) plot of Ip in the presence of PrSSPr for 5 mM (■), 10 mM (●) and 15 mM NaBr (▲). | ||
Fig. 7 displays scans recorded for 15 mM Br− at 10 mV s−1, in the presence of 0 (), 0.15 (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and 1.5 mM (⋯) of PrSSPr, where the presence of the small prepeak is also clearly observed. This behaviour is consistent with a chemical reaction subsequent to the electrogeneration of Br2. The removal of electrogenerated Br2 by chemical reaction makes the initial electron transfer more thermodynamically favourable, and Br2 is produced at lower potentials until the PrSSPr in the vicinity of the electrode is depleted.20,30 As the concentration increased, or if the scan rate was increased above 50 mV s−1 then the two distinct processes merged to form a single observable peak.
) and 1.5 mM (⋯) of PrSSPr, where the presence of the small prepeak is also clearly observed. This behaviour is consistent with a chemical reaction subsequent to the electrogeneration of Br2. The removal of electrogenerated Br2 by chemical reaction makes the initial electron transfer more thermodynamically favourable, and Br2 is produced at lower potentials until the PrSSPr in the vicinity of the electrode is depleted.20,30 As the concentration increased, or if the scan rate was increased above 50 mV s−1 then the two distinct processes merged to form a single observable peak.
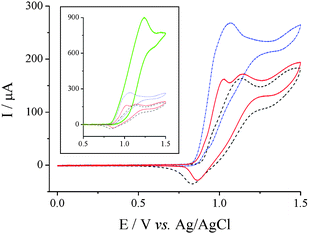 | ||
| Fig. 7
CVs recorded for 15 mM NaBr in 50:50 H2O:AcN (0.1 M NaClO4) at EPPG and 10 mV s−1, in the presence of 0 mM PrSSPr (), 0.15 mM () and 1.5 mM (⋯). The inset displays the same data, with an additional larger peak for 15 mM PrSSPr (). | ||
The inset in Fig. 7 displays the same data overlaid with a scan of 15 mM Br− in the presence of 15 mM PrSSPr. It can be observed that in this case the oxidative current increases significantly, while the reverse peak corresponding to reduction of Br2 is absent. This significant change in oxidative current is consistent with the chemical reaction of Br2 ultimately regenerating Br− in the vicinity of the electrode surface where it is reoxidised again on the time scale of the scan (a so-called catalytic or EC mechanism20). The reaction is relatively rapid, as demonstrated by the complete absence of Br2 on the reverse scan.
The presence of the prepeak on EPPG, and its absence on the SPCE is attributed to the relatively more well-defined, planar geometry of the EPPG facilitating the observation of such relatively small features. In the case of SPCE the two distinct processes (EC and E) merge over the whole range of investigated concentrations, with a resulting simplification of the voltammetry.
In order to confirm the EC mechanism, the CV of 15 mM Br− in the presence and absence of 15 mM PrSSPr at 200 mV s−1 was simulated. A Randles–Ševčík plot derived from a scan rate study gave the bromide diffusion coefficient as 1.06 × 10−5 cm2 s−1, a Tafel plot indicated α ≈ 0.56, while simulation indicated the Br−/Br2 couple possessed E0 ≈ +0.95 V vs.Ag/AgCl and k0 ≈ 0.0005. The reaction of dialkyldisulfides has been reported to go to the alkanesulfonyl bromides,19 which will eventually hydrolyse to form the alkanesulfonic acids,17 ultimately consuming 5 eq. of Br2 per dialkyldisulfide.17,19 The voltammetry in the presence of 15 mM PrSSPr could be simulated using each PrSSPr consuming between 1 to 8 eq. Br2 simply by varying kf; however, for 1 to 4 equivalents of Br2 the peak was relatively sharp due to rapid depletion of the PrSSPr. Satisfactory fits of Ip and the current at E = +1.50 V (the +ve vertex potential) could only be obtained for reactions consuming 5 to 8 eq. Br2, with a value of kf ≈ 375 L mol−1 s−1 required for best fit when 5 eq. Br2 were consumed.
Quantification of the strength of real samples of garlic using SPCE
Two real samples of raw garlic puree were investigated in order to demonstrate proof-of-concept, namely that the quantity of garlic present could be electrochemically quantified using the electrochemistry of NaBr as a probe, and therefore the ‘strength of garlic’ could be inferred. Such techniques have direct implication for the monitoring of garlic during long-term storage, quantifying the ingredients of medicinal garlic supplements, as well as a range of other quality control applications. The garlic samples were sourced from Spain and China; personal communication from the supplier indicated that the Spanish garlic could be classed as ‘strong’ and the Chinese garlic as ‘less strong’.The extraction process used to investigate the garlic was as follows; first a certain amount of garlic was weighed into a 15 mL centrifuge tube, 10 mL of acetonitrile was added, and the tube manually shaken by hand for exactly 5 min. During this process the puree was observed to aggregate to form a slightly desiccated-looking ball, from which the acetonitrile could easily be decanted. For extractions employing large quantities of garlic (>2 g) the neck of the tube was loosely packed with glass wool, the tube inverted and the acetonitrile drained out. This extraction process proceeded smoothly, although presumably other forms of mild agitation, as well as other non-aqueous solvents (such as ethanol) would also be effective.
After extraction 8 mL of the acetonitrile was combined with 8 mL of aqueous solution to form a solution containing 15 mM NaBr and 0.1 M NaClO4. The resulting solutions were investigated voltammetrically, and qualitatively no differences were observed between the garlic-extract samples and those previously investigated containing dipropyldisulfide or diallyldisulfide. Fig. 8 displays an overlay of CVs after extracting various quantities of Spanish garlic. Fig. 9 displays a plot of Ip for the oxidative peak at ca. +1.15 V vs. the quantity of garlic used during the extraction. It can clearly be observed that the stronger Spanish garlic generated a larger signal with increasing weight, corresponding to more dialkyldisulfides present, and clearly demonstrates the potential of this electrochemical technique to rank the garlic samples in order of their strength.
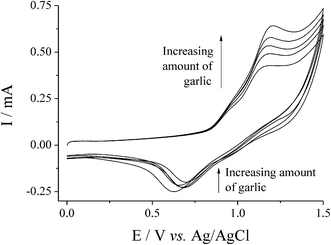 | ||
| Fig. 8
CVs for the oxidation of 15 mM NaBr in 16 mL of 50:50 H2O:AcN (0.1 M NaClO4) at SPCE, where the 8 mL of AcN was first shaken for 5 min with 0, 0.33, 0.67, 1.00 and 1.33 g of Spanish garlic puree. Recorded at 50 mV s−1. | ||
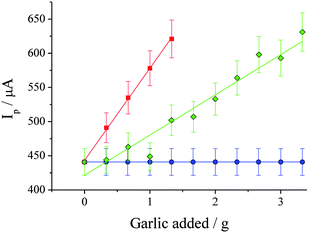 | ||
| Fig. 9 Plot of Ip at ca. +1.15 V vs. weight of garlic used in extraction step (recorded by CV using the parameters specified in Fig. 2), for Spanish garlic (■, gradient = 135 ± 3 µA g−1, R2 = 0.999) and Chinese garlic (◆, gradient = 59 ± 5 µA g−1, R2 = 0.936). Also shown are the values for 15 mM Br− by itself (●). Error bars are based upon measurements in triplicate or more, and include both extraction error and SCPE batch-to-batch variation error. | ||
One extracted sample was investigated using three SPCEs, and the resulting variation in Ip was found to have an error of 1.4%. The extraction process was also repeated separately three times and each solution investigated with a new SPCE, with the combined extraction/SPCE error found to be 4.2%. The error bars in Fig. 9 come from a number of experiments in which the extractions were repeated in triplicate. These experiments were performed over a period of five days, and therefore include day-to-day variation.
By comparing the gradient obtained for diallyldisulfide in Fig. 5 (224 µA mM−1) with those obtained in Fig. 8 for Spanish (135 µA g−1) and Chinese (59 µA g−1) garlic, it can be determined that the extracted disulfide species corresponded to roughly 0.14 and 0.06 w/w% of the garlic puree, respectively. These are reasonable values, with the total weight of the precursor alliin being found between 0.4 and 1.4% w/w in fresh garlic.2
Conclusions
The electrochemistry of bromide at edge plane pyrolytic graphite (EPPG) and screen printed carbon electrodes (SPCEs) in 50:50 water:acetonitrile has been shown to be a highly sensitive probe for dialkyldisulfide concentration. Dipropyldisulfide was detected with an LOD value of 0.067 mM at SPCE. The response of diallyldisulfide was shown to be identical to that of dipropyldisulfide.
Garlic puree was shaken in acetonitrile for 5 min, then the resulting voltammetry of 15 mM bromide recorded using a cheap, disposable SPCE. The voltammetry was able to quantify the dialkyldisulfides extracted into the acetonitrile, providing a proof-of-principle that the strength of the garlic purees can be voltammetrically determined.
Acknowledgements
BCMM would like to thank the EPSRC for a Vacation Bursary. Rob Sweet and Beacon Foods Ltd (Brecon Powys, UK) are gratefully acknowledged for the donation of 2 kg of garlic puree.References
- S. N. Lee, N. S. Kim and D. S. Lee, Anal. Bioanal. Chem., 2003, 377, 749–756 CrossRef CAS.
- E. Block, Garlic and Other Alliums: The Lore and the Science, RSC Publishing, Cambridge, 2010 Search PubMed.
- T. H. Yu, C. M. Wu and C. T. Ho, J. Agric. Food Chem., 1993, 41, 800–805 CrossRef CAS.
- M. E. Rybak, E. M. Calvey and J. M. Harnly, J. Agric. Food Chem., 2004, 52, 682–687 CrossRef CAS.
- S. M. Kim, C. M. Wu, A. Kobayashi, K. Kubota and J. Okumura, J. Agric. Food Chem., 1995, 43, 2951–2955 CrossRef CAS.
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Analyst, 2008, 133, 888–895 RSC.
- P. C. White, N. S. Lawrence, J. Davis and R. G. Compton, Electroanalysis, 2002, 14, 89–98 CrossRef CAS.
- W. Stricks, I. M. Kolthoff and N. Tanaka, Anal. Chem., 1954, 26, 299–303 CrossRef CAS.
- R. Vonwandruszka, X. Yuan and M. J. Morra, Talanta, 1993, 40, 37–42 CrossRef CAS.
- F. G. Banica, A. G. Fogg and J. C. Moreira, Talanta, 1995, 42, 227–234 CrossRef CAS.
- P. J. Vandeberg and D. C. Johnson, Anal. Chim. Acta, 1994, 290, 317–327 CrossRef CAS.
- P. J. Vandeberg and D. C. Johnson, Anal. Chem., 1993, 65, 2713–2718 CrossRef CAS.
- W. R. Lacourse and G. S. Owens, Anal. Chim. Acta, 1995, 307, 301–319 CrossRef CAS.
- G. S. Owens and W. R. LaCourse, J. Chromatogr., B: Biomed. Sci. Appl., 1997, 695, 15–25 CrossRef CAS.
- L. A. Holland and S. M. Lunte, Anal. Chem., 1999, 71, 407–412 CrossRef CAS.
- X. J. Huang and W. T. Kok, J. Liq. Chromatogr., 1991, 14, 2207–2221 CAS.
- G. Sanchezcano, V. Montiel and A. Aldaz, Tetrahedron, 1991, 47, 877–886 CrossRef CAS.
- S. Totebergkaulen and E. Steckhan, Tetrahedron, 1988, 44, 4389–4397 CrossRef CAS.
- S. Siggia and R. L. Edsberg, Anal. Chem., 1948, 20, 938–939 CrossRef CAS.
- R. G. Compton and C. E. Banks, Understanding Voltammetry, World Scientific Publishing Co. Pte. Ltd, Oxford, 2007 Search PubMed.
- N. A. Choudhry, R. O. Kadara, N. Jenkinson and C. E. Banks, Electrochem. Commun., 2010, 12, 406–409 CrossRef CAS.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Electrochem. Commun., 2009, 11, 1377–1380 CrossRef CAS.
- N. D. McGuire, K. C. Honeychurch and J. P. Hart, Electroanalysis, 2009, 21, 2165–2170 CrossRef.
- K. C. Honeychurch, L. Gilbert and J. P. Hart, Anal. Bioanal. Chem., 2010, 396, 3103–3111 CrossRef CAS.
- G. D. Allen, M. C. Buzzeo, L. G. Davies, C. Villagran, C. Hardacre and R. G. Compton, J. Phys. Chem. B, 2004, 108, 16322–16327 CrossRef CAS.
- G. D. Allen, M. C. Buzzeo, C. Villagran, C. Hardacre and R. G. Compton, J. Electroanal. Chem., 2005, 575, 311–320 CrossRef CAS.
- P. Fanjul-Bolado, D. Hernandez-Santos, P. J. Lamas-Ardisana, A. Martin-Pernia and A. Costa-Garcia, Electrochim. Acta, 2008, 53, 3635–3642 CrossRef CAS.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Sens. Actuators, B, 2009, 138, 556–562 CrossRef.
- C. M. A. Brett and A. M. O. Brett, Electroanalysis, Oxford University Press, Oxford, 1998 Search PubMed.
- T. Matsue, H. Yamada, H. C. Chang and I. Uchida, Bioelectrochem. Bioenerg., 1990, 24, 347–354 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |