A method for N-terminal de novo sequencing of Nα-blocked proteins by mass spectrometry
Chihiro
Nakajima
ab,
Hiroki
Kuyama
*a,
Takashi
Nakazawa
c,
Osamu
Nishimura
a and
Susumu
Tsunasawa
a
aInstitute for Protein Research, Osaka University, Osaka, 565-0871, Japan. E-mail: kuyama@protein.osaka-u.ac.jp
bLife Science Research Center, Shimadzu Corporation, Kyoto, 619-0237, Japan
cDepartment of Chemistry, Nara Women's University, Nara, 630-8506, Japan
First published on 8th October 2010
Abstract
A method for de novo sequencing of Nα-blocked proteins by mass spectrometry (MS) is presented. The approach consists of enzymatic digestion of Nα-blocked protein, recovery of N-terminal peptide by depletion of non-N-terminal peptides from the digest pool, and selective derivatization of a C-terminal α-carboxyl group of isolated N-terminal peptide. The C-terminal α-carboxyl group of the N-terminal peptide was selectively derivatized with 3-aminopropyl-tris(2,4,6-trimethoxyphenyl)phosphonium bromide (TMPP-propylamine), according to oxazolone chemistry. The reagent TMPP-propylamine was designed to facilitate sequence analysis with MALDI-MS by mass- and charge-tagging. All of the identities and N-terminal sequences of two Nα-acetylated proteins (rabbit phosphorylase b and bovine calmodulin) and human orexin A, which has pyroglutamic acid at the N-terminus, were successfully analyzed by allowing for the y-type ions almost exclusively.
Introduction
Post-translational modifications (PTMs) of proteins occur to accomplish their maturation and are key processes for exhibiting the requisite biological functions. It is therefore essential to characterize each protein by determining the identity of its PTM as well as its amino acid sequence. Among PTMs, N-terminal acetylation is a widespread modification in eukaryotes. The estimated extent of mature proteins with N-terminal acetylation varies widely from 40 to 50% for yeast proteins and up to 90% for higher eukaryote proteins.1 Unlike other forms of PTMs, the acetylation of N-terminal amino groups imposes a great difficulty on protein sequencing, the majority of which relies on the reactivity of the free amino group to various chemical reagents.Mass spectrometry (MS) has had substantial success in the sequencing of terminally blocked peptides, especially when it is employed in combination with a highly efficient scheme for separating or isolating the terminal peptides. The suggested approaches for analyzing N-terminally blocked peptides include those based on enriching the target peptides from peptides generated by proteolytic cleavage.2–9 The most impressive realization of this idea has been reported in connection with ‘positional proteomics’, which exploits the enrichment of Nα-acetylated peptides using an amine-reactive immobilized reagent.2,6–9 However, the absence of a free α-amino group in a peptide thus enriched often impedes the access of the chemical reagents generally used in the sequencing strategies of MS to enhance the sensitivity or simplify the spectra. In principle, every Nα-blocked peptide still retains a C-terminal carboxyl group free to be modified, albeit to little avail because of the difficulty in activating it specifically and effectively for chemical derivatization.
In earlier studies, we developed a method for isolating the N-terminal peptides of proteins using p-phenylenediisothiocyanate (DITC) glass from proteolytic digest.8,9 We have also devised a scheme for selectively modifying C-terminal α-carboxyl groups based on oxazolone chemistry.10,11 An oxazolone forms specifically at the C-termini of peptides, so that the C-terminal carboxyl group can be strictly distinguished from those in the side-chains of glutamic acid and aspartic acid. However, the reaction yields were not high enough to envisage applying this to the sequence analysis of isolated terminal peptides at sub-nanomolar quantities, until the successful optimization of the reaction conditions has been achieved.12 On the technical grounds elaborated so far, we suggest here a method for sequencing Nα-blocked peptides (Fig. 1). The procedures include: (1) proteolytic digestion of Nα-blocked proteins with peptidyl-Lys metalloendopeptidase (LysN),13–15 (2) isolation of terminal Nα-blocked peptides from the enzyme digest by removing all of the peptides having free amino groups with the DITC glass,16 and (3) selective activation of the C-terminal α-carboxyl group for amidation with 3-aminopropyl-tris(2,4,6-trimethoxyphenyl)phosphonium bromide (TMPP-propylamine17,18), which might have the effect of preferentially giving a defined set of fragment peaks in MALDI-TOF/TOF-MS, thus facilitating the amino acid sequencing. We chose to tag the peptides with the TMPP group, which has already proved useful for de novo sequencing as demonstrated in the previous studies, where it was utilized as a tag for the α-amino group.8,19–21
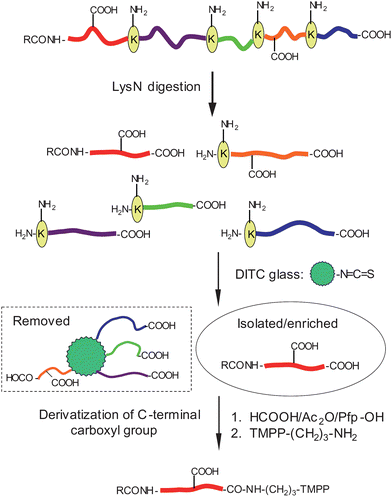 | ||
| Fig. 1 Schematic depiction of this protocol. | ||
In this paper we will also present an extension of the method, adapted to the case where the use of LysN digestion to obtain the N-blocked peptide is unsuccessful. A discussion about the wider applicability of modifying α-carboxyl groups with TMPP-propylamine in proteomics studies based on mass spectrometry is included.
Experimental
Materials
Ac-SQNY was purchased from American Peptide Company (Sunnyvale, CA, USA). Phosphorylase b (rabbit), orexin A (human), calmodulin (bovine), tris(2,4,6-trimethoxyphenyl)phosphine (TMPP), acetic anhydride (Ac2O), 3-bromopropylamine hydrochloride, and iodoacetamide were obtained from Sigma-Aldrich (St. Louis, MO, USA). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was obtained from Fluka (Switzerland). Peptidyl-Lys metalloendopeptidase (LysN) from Grifola frondosa was purchased from Seikagaku Co. (Tokyo, Japan). Acetonitrile (ACN), 2-propanol, formic acid, pentafluorophenol (Pfp-OH), potassium carbonate, toluene, sodium chloride, magnesium sulfate, diethyl ether, and trifluoroacetic acid (TFA) were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). α-Cyano-4-hydroxycinnamic acid (CHCA: high-purity mass-spectrometric grade) was obtained from Shimadzu GLC (Tokyo, Japan). The DITC glass was prepared in-house according to the method of Wachter et al.,16 using aminopropyl glass (average pore size 170 Å, 200 to −400 mesh; amine content 162 μmol/g) and 1,4-phenylenediisothiocyanate obtained from Sigma. Methanediphosphonic acid (MDPNA) was obtained from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). The water used in all of the experiments was purified using a MilliQ water purification system. All other chemicals were of analytical reagent grade and were used without further purification.Preparation of 3-aminopropyl-tris(2,4,6-trimethoxyphenyl)phosphonium bromide (TMPP-propylamine)
TMPP-propylamine was prepared according to the reported procedure.17,18 A solution of 3-bromopropylamine was prepared by mixing 3-bromopropylamine hydrochloride (160 mg; 0.73 mmol) and potassium carbonate (260 mg) in water (2 mL), to which toluene (2 mL) was added to extract the free base. The toluene layer was washed with brine and dried over magnesium sulfate. To the toluene solution was added tris(2,4,6-trimethoxyphenyl)phosphine (200 mg; 0.38 mmol), and the solution was heated at 85 °C for 5 h, during which the desired TMPP-propylamine bromide precipitated as a white powder. The reaction product was collected by filtration, washed with toluene (3 × 1 mL) and diethyl ether (3 mL), and dried for 12 h in vacuo. The yield of the obtained white powder was 110 mg (51%). MS analysis indicated that it was a sole product. And the structure was confirmed also by MS. This reagent can be stored for at least one year under −30 °C.Digestion with LysN protease and derivatization of α-carboxyl groups using TMPP-propylamine: the case of phosphorylase b and orexin A
N-terminal peptides devoid of the amino group were obtained from proteins (100 pmol each) by a protocol using LysN digestion and DITC treatment, as reported previously.7 The N-terminal peptide isolated from the proteolytic digest was purified with ZipTip, followed by vacuum-centrifuge drying. The N-terminal peptide was re-dissolved in 12 μL of a HCOOH–Ac2O–Pfp-OH mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in volume), and incubated for 20 min at 60 °C. After the liquid materials were removed using a vacuum evaporator, the remaining residue was mixed with 14 μL of a 5:2 mixture of toluene and Pfp-OH and evaporated again.12 Two microliters of aqueous TMPP-propylamine solution (5 nmol/μL) and 2 μL of Et3N–MeOH (1:1, v/v) were added to the activated peptide and the mixture was subsequently sonicated for 10 min and then left standing for 20 min. For mass analysis, the reaction mixture was diluted with water (26 μL) and an aliquot (0.4 μL) was used.
:1:1 in volume), and incubated for 20 min at 60 °C. After the liquid materials were removed using a vacuum evaporator, the remaining residue was mixed with 14 μL of a 5:2 mixture of toluene and Pfp-OH and evaporated again.12 Two microliters of aqueous TMPP-propylamine solution (5 nmol/μL) and 2 μL of Et3N–MeOH (1:1, v/v) were added to the activated peptide and the mixture was subsequently sonicated for 10 min and then left standing for 20 min. For mass analysis, the reaction mixture was diluted with water (26 μL) and an aliquot (0.4 μL) was used.
Digestion with trypsin and guanidination of lysine residues: the case of calmodulin
Calmodulin, with which proteolytic digestion with LysN was inefficient for recovering N-terminally blocked peptide, was first digested with trypsin. The ε-amino group of lysine residues was guanidinated with O-methylisourea.22 The calmodulin (100 pmol) was dissolved with 3 μL of urea solution (8 M in 50 mM NaHCO3), which was then diluted with 30 μL of aqueous NaHCO3 (50 mM) and 2 μL of ACN. To the solution, 4 μL of aqueous trypsin solution (50 ng/μL) was added and incubated at 37 °C for 16 h. The trypsin digest was evaporated to dryness by vacuum centrifuge and an aqueous solution of O-methylisourea (5 μL, 0.5 M) and aqueous ammonia (20 μL, 7 M) was added for the guanidination, conducted at 60 °C for 1 h. After concentration using a vacuum centrifuge, the peptide was diluted with 0.5% TFA (20 μL) and the solution was treated with ZipTip to remove salts and reagents. The peptide eluted from ZipTip was then evaporated with a vacuum centrifuge and re-dissolved in 100 μL of aqueous NaHCO3 (50 mM) and 10 μL of ACN. The N-terminal peptide was collected using the reported procedure, and the downstream operations were the same as those for phosphorylase b and orexin A.MALDI-TOF MS
MALDI mass spectra were recorded on an AXIMA CFR-plus (Shimadzu/Kratos, Manchester, UK) reflectron time-of-flight mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse width). All measurements were performed in the positive-ion reflectron mode. The ion acceleration voltage was set to 20 kV, and the reflectron detector was operated at 24 kV. The flight path in the reflectron mode is 240 cm. PSD mode was used for the MS/MS experiments.The matrix used in this experiment was α-cyano-4-hydroxycinnamic acid (CHCA), which was dissolved to saturation in 50% aqueous ACN containing 0.05% TFA. We used MDPNA, which has been proven useful for MALDI analysis of salt-containing samples, as a matrix additive.23 MDPNA was used as 1% aqueous solution. An aliquot (0.4 μL) of sample solution was mixed with an equivalent volume of matrix and matrix-additive solutions on the MALDI target plate and analyzed after drying.
The m/z values in the spectra were externally calibrated with angiotensin II (human) and ACTH fragment 18–39 (human) using CHCA as a matrix.
Results and discussion
Outline of the derivatization of α-carboxyl group
In our study pursuing a mass spectrometry-based methodology to clarify the structure of the mature protein, in particular de novo sequencing of the protein's terminal peptides, we reported some methods and applications. These all involve a free α-amino group which is derivatized by 3-sulfopropionylation9 or TMPP-acetylation8,21 for enabling de novo sequencing by MS. Therefore, Nα-blocked protein, which is devoid of a free α-amino group, cannot be derivatized with mass-tag reagents for de novo sequencing.Hence, we focused on employing the α-carboxyl group, another characteristic group of a protein or peptide, for derivatizing N-terminal peptide isolated from Nα-blocked protein.
In this study, we used specific activation of an α-carboxyl group through oxazolone formation and TMPP-propylamine as a derivatizing agent (Fig. 1) because TMPP-acetylation of the α-amino group of a peptide is known to provide better fragmentation for de novo sequence analysis by MS.
Oxazolone formation is a well-known characteristic reaction with an α-carboxyl group. Oxazolone is a five-membered ring selectively formed between an α-carboxyl group and a carbonyl group of a penultimate amino-acid residue from the C-terminus (Fig. 2), and serves as a reactive electrophile for reactions with various types of nucleophiles. In a previous report,12 we described the methyl amidation of an α-carboxyl group with high selectivity. In this paper, we applied the protocol to the selective derivatization of an α-carboxyl group using TMPP-propylamine (Fig. 2).
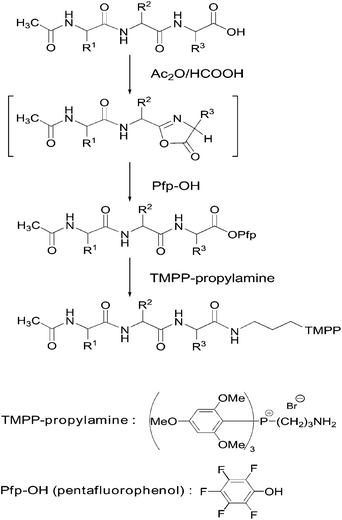 | ||
| Fig. 2 Outline of α-carboxyl group derivatization using TMPP-propylamine. | ||
Model peptide experiment
We used a model peptide, Ac-SQNY, to test the selective C-terminal derivatization using the protocol reported previously.12 A hundred picomoles of the model peptide was derivatized with TMPP-propylamine, where the molar ratio of peptide:reagent was 1:20. As the spectrum shows, the peptide derivatized with TMPP-propylamine was detected as almost the sole peak at m/z 1124.6 (Fig. 3a). Apparently, side reactions such as the modification of side-chain hydroxyl and the amide groups on it did not occur under the present reaction conditions. The identity of the TMPP-derivatized peptide was confirmed by an MS/MS analysis in which y-type ions were observed preferentially, making it easier to obtain sequence information (Fig. 3b). The preference for this fragmentation pattern is in sharp contrast to that of the N-terminally TMPP-acetylated peptides, which tend to give a-type, but not b-type ions.19,20
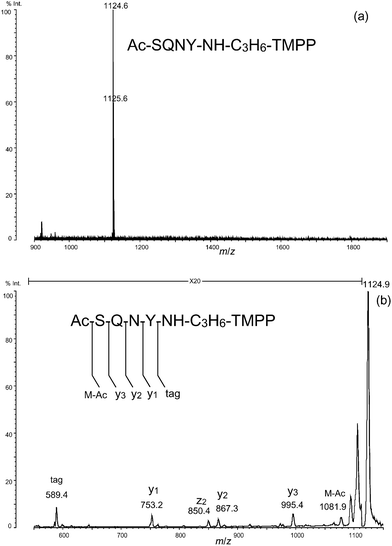 | ||
| Fig. 3 (a) Derivatization of Ac-SQNY with TMPP-propylamine. (b) MS/MS spectrum of the derivatized peptide. | ||
It has been estimated from the peak intensities in MALDI-MS that 60–90% of the C-terminal carboxyl group underwent amidation with methylamine through the oxazolone.12 Although we have not determined the yield of the reaction with TMPP-propylamine, it would be somewhat lower because the concentration of the amine used was about 2.5 mM, much lower than the 0.5–1 M for methylamine. Nevertheless, the possible low yield of the derivatization with TMPP-propylamine could have been successfully compensated for by a sensitivity-enhancing effect of the TMPP group, thus apparently suppressing the detection of undesirable peaks including those of unmodified N-terminal peptides. This is consistent with the tendency to detect a-type (y-type) ions when the TMPP group is attached to an N-terminus (C-terminus) in MS/MS experiments.
Experiments with model proteins
To test the feasibility of the protocol illustrated in Fig. 1, including the C-terminal derivatization for sequence analysis by MS, we chose three protein models: rabbit phosphorylase b, human orexin A, and bovine calmodulin from commercially available Nα-blocked proteins. Phosphorylase b and calmodulin are Nα-acetylated, and orexin A has pyroglutamic acid residue (Pyr) at the N-terminus.As judged from the peaks at m/z 973.4 for phosphorylase b and m/z 1156.6 for orexin A, and in agreement with the mass values expected from their amino-acid sequences and N-terminal structures, N-terminal peptides of these proteins were isolated by the reported protocol7 using metalloendoprotease LysN digestion and DITC glass (Fig. 4a and 5a). The simplicity of the spectra suggests that the isolation procedure of these N-terminal peptides using DITC glass has effectively removed other internal and C-terminal peptides. Due to the derivatization of C-termini with TMPP-propylamine, the mass of each peptide increased by 572 Da, measured by the shift in the corresponding peaks to m/z 1544.9 for phosphorylase b and m/z 1727.7 for orexin A (Fig. 4b and 5b).
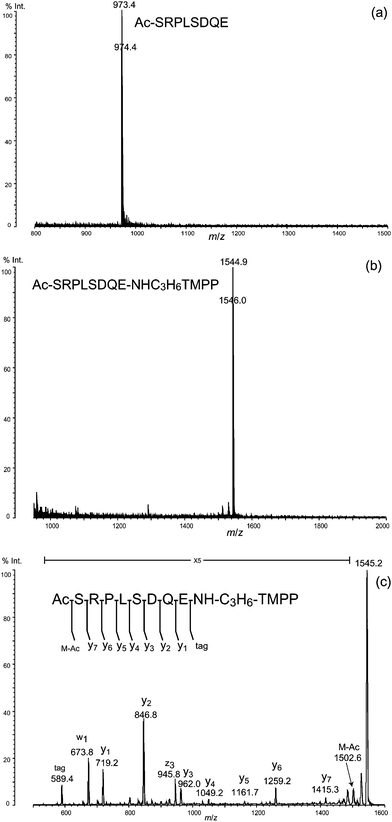 | ||
| Fig. 4 (a) Isolated N-terminal peptide of phosphorylase b (rabbit). (b) Derivatization of the N-terminal peptide with TMPP-propylamine. (c) MS/MS spectrum of the derivatized N-terminal peptide. | ||
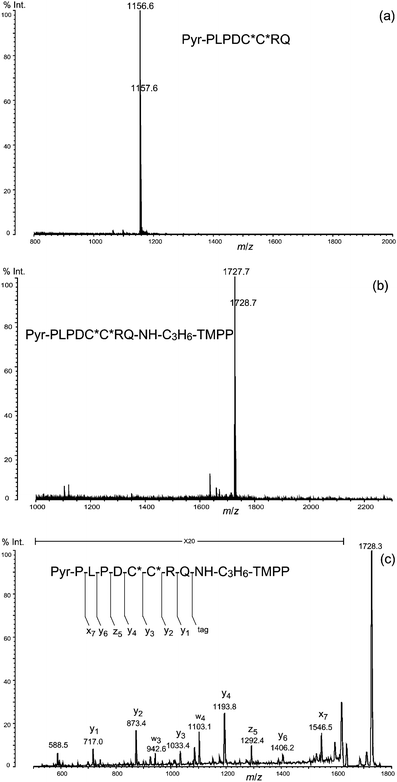 | ||
| Fig. 5 (a) Isolated N-terminal peptide of orexin A (human). (b) Derivatization of the N-terminal peptide with TMPP-propylamine. (c) MS/MS spectrum of the derivatized N-terminal peptide. C*: S-carbamidomethyl cysteine residue; Pyr: pyroglutamic acid residue. | ||
We noted that the terminal Nα-acetylated peptide of calmodulin was not obtained by this protocol, probably because of a mis-cleavage along peptide bond, say Phe12–Lys13 in the N-terminal peptide during digestion with LysN, leaving an ε-amino group of lysine residue (Lys13) that could react with an isothiocyanate group in the DITC glass. In fact, the N-terminal peptide, Ac-ADQLTEEQIAEFJ (J indicates homoarginine) was isolated from calmodulin after digestion with trypsin used in place of LysN, guanidination of the ε-amino group of Lys residues, and treatment with DITC glass. The peptide thus isolated gave a peak at m/z 1605.8 (Fig. 6a), which shifted to m/z 2177.2 with an increase in the mass of 572 Da, characteristic of derivatization with TMPP-propylamine (Fig. 6b). The PSD spectrum of the peak at m/z 2177.2 (Fig. 6c) clearly indicates that the derivatization occurred exclusively at the C-terminal carboxyl group in the presence of four side-chain carboxyl groups of aspartic acid (Asp2) and glutamic acid (Glu6, Glu7, and Glu11), in agreement with the results of preceding experiments conducted on the basis of oxazolone chemistry.10–12 Furthermore, a series of y-type ions appeared predominantly in the PSD spectrum, making it very easy to clarify the amino acid sequence. Note that the preference for y-type ions was observed commonly in the MS/MS spectra for peptides attached with the TMPP group at the C-termini (Fig. 4c, 5c, and 6c). This is in sharp contrast to the tendency of observed a-type ions in the fragmentation of peptides modified with the TMPP-acetyl group at the N-termini.8,19–21 Unlike Nα-TMPP-acetylated peptides in which the N-terminal arginine residue causes deterioration in the appearance of the MS/MS spectra,24 the modification of the C-terminal arginine residue with TMPP-propylamine had no such damaging effect on the fragmentation pattern of the peptide (see, for example, Fig. 4c and 5c). If need be, however, derivatization of arginine with acetylacetone to N5-(4,6-dimethyl-2-pyrimidinyl)ornithine (Pyo)24,25 (Fig. 7a) could be useful for further simplifying the MS/MS spectrum, as we demonstrated by showing that the peak of the x7-ion, potentially confusing the MS/MS spectrum (Fig. 5c) was converted to that of the desired y7-ion to complete a set of y-type ions (Fig. 7b).
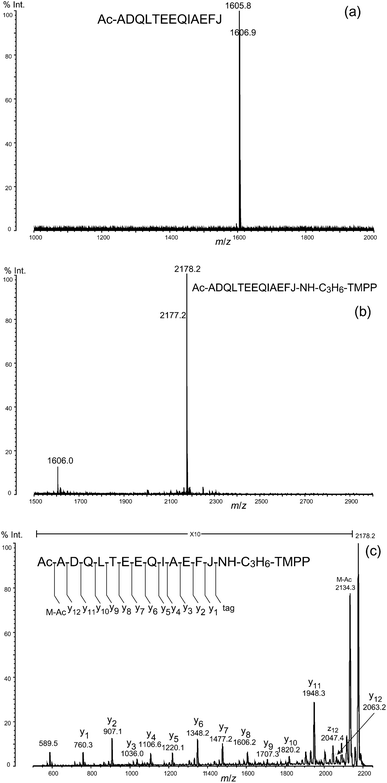 | ||
| Fig. 6 (a) Isolated N-terminal peptide of calmodulin (bovine). (b) Derivatization of the N-terminal peptide with TMPP-propylamine. (c) MS/MS spectrum of the derivatized N-terminal peptide J: homoarginine residue. | ||
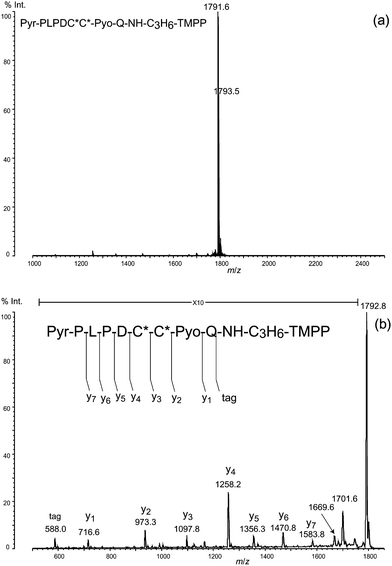 | ||
| Fig. 7 Simplifying fragmentation by converting Arg to Pyo residue. (a) Arg derivatization using acetylacetone to Pyo residue. (b) MS/MS spectrum of the arginine-derivatized N-terminal peptide. Pyo: N5-(4,6-dimethyl-2-pyrimidinyl)ornithine residue; C*: S-carbamidomethyl cysteine residue. | ||
Other fragmentation patterns characteristic of the peptides modified with TMPP-propylamine at the C-termini include the observation of z-type ions coupled with y-type ions, both of which occur due to fragmentation on the N-terminal side of aspartic acid. In this pair, the peaks of z-type ions are often more intense than those of y-type ions (Fig. 4c, 5c, and 6c). Another pair of peaks representing y-type and w-type (y-SCH2CONH2) ions was observed at a comparable intensity, suggesting that the y-type ion produced by the cleavage of the bond at the N-terminal side of (S-carboxamidomethyl)cysteine is likely to undergo β-elimination (Fig. 5c). Although an x-type ion corresponding to fragmentation at the bond of Pro1–Leu2 was detected in the MS/MS of the N-terminal peptide from orexin A (Fig. 5c), this might not be a common feature of bond breaking where proline or leucine residue is involved because no peak of an x-type ion appeared in the PSD spectrum of the N-terminal peptide of phosphorylase b. Instead, a peak of a y5-ion, signifying fragmentation in Pro4–Leu5, was clearly observed. For all of these observations of unpredicted peaks, the C-terminal TMPP-propylamide group could effectively simplify MS/MS spectra by providing an almost complete set of peaks of y-type ions, thus making it easier to determine the amino-acid sequence.
One of the distinguishing features of C-terminal TMPP-propylamidation is the simplicity of the fragmentation pattern in MS/MS that exhibits a complete or near complete y-type ion series. This greatly facilitates the de novo sequencing of peptides. The cleanliness of the reaction is such that no peak was observed at a mass exceeding the expected value by 28 Da, corresponding to formylation of the hydroxyl group of a serine or threonine residue.
It is well known that in using a TMPP-Ac charge tag, sample clean up to remove excess reagent is necessary prior to MALDI-MS analysis.26,27 As described in the previous report,8 we used MDPNA23 as a co-matrix for the detection of TMPP-acetylated peptide. Just an addition of the reagent as a co-matrix improved the S/N ratio of the derivatized peptide peaks. This is an advantage where conventional ZipTip pretreatment is often not effective. And if need be, the excess reagent in the C-terminal TMPP-propylamidation can be removed by an appropriate amine-reactive medium such as DITC glass, which does not capture the peptide(s) of interest because of the lack of the free amine group.
Concerning C-terminal carboxyl derivatization in connection with de novo sequencing of peptides, Lindh et al.28 employed a condensing reagent (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride: EDC) for coupling with 4-aminonaphthalenesulfonic acid as a charge tag. With this approach, however, it is inherently impossible to distinguish between the C-terminal and side-chain carboxyl groups. Our method, requiring a strict distinction between these two types of carboxyl groups, is incompatible with the non-specific nature of this simple condensation scheme. Note that satisfactory performance of the present method has been achieved through restricting the position of charge tagging to the C-termini of peptides, so that an almost complete set of peaks due to y-type ions could be observed in the MS/MS spectra.
Prospects for future improvements
Choosing a proteolytic enzyme appropriate for isolating N-terminally blocked peptide(s) is a key issue in the present method. We employed LysN protease because it can specifically cleave peptide bonds involving X–Lys, leaving the N-terminal peptide of interest free of amino groups. However, not all of the X–Lys bonds are cleaved by LysN protease at a comparable rate; those known to be difficult to cleave include Pro–Lys and X(an acidic residue)–Lys.7,13,14 Actually, the N-terminal peptide of calmodulin was not obtained by the original protocol utilizing the proteolysis with LysN (Fig. 1), probably because a mis-cleavage in the peptide bond Phe12–Lys13 left a free ε-amino group of lysine in the peptide, ensuring its undesirable capture by the DITC glass. Although we have managed to solve this problem by using trypsin followed by protection of the ε-amino group by guanidination, any other endoproteinase with a defined specificity can be employed in place of LysN provided that proteolysis does not leave a free amino group in an N-terminal peptide of interest. Otherwise, the free amino group in the N-terminal peptide should be protected while leaving the α-amino groups of other peptides free of protection. In this regard, guanidination of the ε-amino group with O-methylisourea satisfies the latter criterion, which requires that the modification be specific to the ε-amino group. Additionally, the conversion of lysine to homoarginine (J) is associated with an increase in mass by 170 Da, making it possible to differentiate lysine and glutamine residues, which are isobaric (residue mass of 128 Da). A slight extension of this procedure involving guanidination could allow the use of other endopeptidases such as GluC, AspN, or chymotrypsin for preparing N-terminally blocked peptides.Conclusion
De novo sequence analysis of N-terminal peptide of Nα-blocked protein was successfully performed by employing a combination of isolation of the N-terminal peptide from protease digest and highly selective derivatization of an α-carboxyl group using 3-aminopropyl-tris(2,4,6-trimethoxyphenyl)phosphonium bromide.The oxazolone reaction used for the introduction of the TMPP-propylamine is site-specific for the C-terminus, and the side chain carboxyl group is not derivatized. The TMPP group is a good tag for MALDI, because it increases the S/N ratio and gives a high quality PSD spectrum in MS analysis, often with a complete or near complete y-type ion series when attached to the C-terminus, and the simplified ion series can be used directly to interpret the amino-acid sequence. The fragmentation pattern differs from that of the peptide incorporating the TMPP-acetyl group at its α-amino group, which includes mainly a-type ions.
Acknowledgements
This study was supported in part by a Grant-in-Aid for Scientific Research (C) (Grant 22510230 to H.K.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.References
- B. Polevoda and F. Sherman, J. Biol. Chem., 2000, 275, 36479–36482 CrossRef CAS.
- T. H. Akiyama, T. Sasagawa, M. Suzuki and K. Titani, Anal. Biochem., 1994, 222, 210–216 CrossRef CAS.
- K. Gevaert, M. Goethals, L. Martens, J. Van Damme, A. States, G. R. Thomas and J. Vandekerckhove, Nat. Biotechnol., 2003, 21, 566–569 CrossRef CAS.
- K. Kuhn, A. Thompson, T. Prinz, J. Muller, C. Baumann, G. Schmidt, T. Neumann and C. Hamon, J. Proteome Res., 2003, 2, 598–609 CrossRef CAS.
- L. McDonald, D. H. Robertson, J. L. Hurst and R. J. Beynon, Nat. Methods, 2005, 2, 955–957 CrossRef CAS.
- T. Mikami and T. Takao, Anal. Chem., 2007, 79, 7910–7915 CrossRef CAS.
- G. Coussot, D. H. Hawke, A. Mulartz, J. H. Koomen and R. Kobayashi, Anal. Biochem., 2007, 361, 302–304 CrossRef CAS.
- H. Kuyama, K. Sonomura, O. Nishimura and S. Tsunasawa, Anal. Biochem., 2008, 380, 291–296 CrossRef CAS.
- M. Yamaguchi, D. Nakayama, K. Shima, H. Kuyama, E. Ando, T. Okamura, N. Ueyama, T. Nakazawa, S. Norioka, O. Nishimura and S. Tsunasawa, Rapid Commun. Mass Spectrom., 2008, 22, 3313–3319 CrossRef CAS.
- T. Nakazawa, M. Yamaguchi, K. Nishida, H. Kuyama, T. Obama, E. Ando, T. Okamura, N. Ueyama, K. Tanaka and S. Tsunasawa, Rapid Commun. Mass Spectrom., 2004, 18, 799–807 CrossRef CAS.
- M. Yamaguchi, M. Oka, K. Nishida, M. Ishida, A. Hamazaki, H. Kuyama, E. Ando, T. Okamura, N. Ueyama, S. Norioka, O. Nishimura, S. Tsunasawa and T. Nakazawa, Anal. Chem., 2006, 78, 7861–7869 CrossRef CAS.
- H. Kuyama, C. Nakajima, T. Nakazawa, O. Nishimura and S. Tsunasawa, Proteomics, 2009, 9, 4063–4070 CrossRef CAS.
- T. Nonaka, H. Ishikawa, Y. Tsumuraya, Y. Hashimoto, N. Dohmae and K. Takio, J. Biochem., 1995, 118, 1014–1020 CAS.
- T. Nonaka, Y. Hashimoto and K. Takio, J. Biochem., 1998, 124, 157–162 CAS.
- T. Hori, T. Kumasaka, M. Yamamoto, T. Nonaka, N. Tanaka, Y. Hashimoto, T. Ueki and K. Takio, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 361–368 CrossRef CAS.
- E. Wachter, W. Machleidt, H. Hofner and J. Otto, FEBS Lett., 1973, 35, 97–102 CrossRef CAS.
- W. J. Leavens, S. J. Lane, R. M. Carr, A. M. Lockie and I. Waterhouse, Rapid Commun. Mass Spectrom., 2002, 16, 433–441 CrossRef CAS.
- A. J. Cartwright, P. Jones, J.-C. Wolff and E. H. Evans, J. Anal. At. Spectrom., 2005, 20, 75–80 RSC.
- Z. H. Huang, J. Wu, K. D. W. Roth, Y. Yang, D. A. Gage and J. T. Watson, Anal. Chem., 1997, 69, 137–144 CrossRef CAS.
- Z. H. Huang, T. Shen, J. Wu, D. A. Gage and J. T. Watson, Anal. Biochem., 1999, 268, 305–317 CrossRef CAS.
- H. Kuyama, K. Shima, K. Sonomura, M. Yamaguchi, E. Ando, O. Nishimura and S. Tsunasawa, Proteomics, 2008, 8, 1539–1550 CrossRef CAS.
- R. L. Beardsley and J. P. Reilly, Anal. Chem., 2002, 74, 1884–1890 CrossRef CAS.
- H. Kuyama, K. Sonomura and O. Nishimura, Rapid Commun. Mass Spectrom., 2008, 22, 1109–1116 CrossRef CAS.
- H. Kuyama, K. Sonomura, K. Shima, O. Nishimura and S. Tsunasawa, Rapid Commun. Mass Spectrom., 2008, 22, 2063–2072 CrossRef CAS.
- S. Dikler, J. W. Kelly and D. H. Russell, J. Mass Spectrom., 1997, 32, 1337–1349 CrossRef CAS.
- S. Gallien, E. Perrodou, C. Carapito, C. Deshayes, J.-M. Reyrat, A. V. Dorsselaer, O. Poch, C. Schaeffer and O. Lecompte, Genome Res., 2009, 19, 128–135 CAS.
- M. An, J. Dai, Q. Wang, Y. Tong and J. Ji, Rapid Commun. Mass Spectrom., 2010, 24, 1869–1874 CrossRef CAS.
- I. Lindh, L. Hjelmqvist, T. Bergman, J. Sjovall and W. J. Griffiths, J. Am. Soc. Mass Spectrom., 2000, 11, 673–686 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |