Nanomaterial surface chemistry design for advancements in capillary electrophoresis modes
Michael R.
Ivanov
and
Amanda J.
Haes
*
University of Iowa, Department of Chemistry, 204 IATL, Iowa City, Iowa 52242, USA. E-mail: amanda-haes@uiowa.edu; Fax: +1 319-335-1270; Tel: +1 319-384-3695
First published on 22nd October 2010
Abstract
Tailored surface chemistry impacts nanomaterial function and stability in applications including in various capillary electrophoresis (CE) modes. Although colloidal nanoparticles were first integrated as colouring agents in artwork and pottery over 2000 years ago, recent developments in nanoparticle synthesis and surface modification increased their usefulness and incorporation in separation science. For instance, precise control of surface chemistry is critically important in modulating nanoparticle functionality and stability in dynamic environments. Herein, recent developments in nanomaterial pseudostationary and stationary phases will be summarized. First, nanomaterial core and surface chemistry compositions will be classified. Next, characterization methods will be described and related to nanomaterial function in various CE modes. Third, methods and implications of nanomaterial incorporation into CE will be discussed. Finally, nanoparticle-specific mechanisms likely involved in CE will be related to nanomaterial surface chemistry. Better understanding of surface chemistry will improve nanoparticle design for the integration into separation techniques.
 Michael R. Ivanov | Michael R. Ivanov earned a BS in Chemistry from Loras College in Dubuque, Iowa and is presently a PhD candidate in Chemistry at the University of Iowa under the direction of Amanda J. Haes. His research interests include characterization of nanomaterial surface chemistry and investigations into gold nanoparticles applications in capillary electrophoresis. |
 Amanda J. Haes | Amanda J. Haes is currently an Assistant Professor in the Chemistry Department at the University of Iowa. She completed her PhD in Chemistry at Northwestern University with Richard P. Van Duyne. Before beginning her independent career, she was a National Research Council Research Associate with Greg E. Collins at the US Naval Research Laboratory. Haes group research activities include designing and purifying novel nanomaterials for stable spectroscopic studies, improving detection limits of biological and environmental pathogens, and investigating how surface chemistry impacts nanomaterial function in separations and spectroscopy. |
1. Introduction
Capillary electrophoresis (CE)1–3 is an effective technique for the liquid-phase separation of molecules ranging from metal ions4 to biomolecules such as DNA.5,6 CE separation modes7 utilize small sample volumes (nanolitre injection volumes)8 and high separation selectivities.9 Electrically driven capillary separations are achieved by applying a potential to charged molecules (ions) suspended in a solution. Electrophoretic separation of charged species occurs because (1) anions and cations migrate in opposite directions toward electrodes of opposite charge and (2) similarly charged ions with varying Stokes radii have different migration velocities.10To separate neutral species, both pseudostationary (i.e. matrices which can either co-migrate with or migrate against the mobile phase) and stationary phases (i.e. non-moving matrices) are implemented to improve separation selectivity.11 Terabe et al.12,13 pioneered micellar electrokinetic chromatography (MEKC), a technique which uses surfactant micelle additives as a pseudostationary phase to improve the separation of neutral or like charged molecules. Above the critical micelle concentration (CMC), surfactant molecules form micelles with non-polar cores which are protected from the aqueous, mobile phase environment by polar head groups. Because neutral molecules partition between the micelle and mobile phases at different rates, separation selectivity improves vs. traditional CE.
Unfortunately, some hydrophobic molecules can irreversibly partition into micelles thereby reducing separation selectivity and detection specificity.14 As a result, additional organic modifiers can be added to the run buffer to decrease micelle–molecule affinity15–17 or to change the separation mechanism.18 Further efforts to improve CE selectivity are summarized in Fig. 1 and include capillary electrochromatography (CEC),19 capillary gel electrochromatography (CGE),20 and various capillary coatings.21CEC, for instance, combines the high efficiency of an electrophoretic CE separation with the selectivity of HPLC.22,23
 | ||
| Fig. 1 Classification of common capillary electrophoresis modes. | ||
Pseudostationary or stationary phases in these separation techniques can include nanomaterials to further improve selectivity.24,25 Nanomaterials possess ideal properties for integration into CE as pseudostationary and stationary phases. For instance, these materials are inherently “small”—that is, contain at least one dimension less than 100 nm and can be included at low concentrations compared to traditional pseudostationary or stationary phases so that less than 1% of the total capillary volume is occupied by nanoparticles.26 Second, nanomaterials exhibit inherently large surface area to volume ratios and novel size dependent chemical and physical properties.27,28
Nanomaterials have been used in CE separations for over two decades. In 1989, Wallingford and Ewing demonstrated that a pseudostationary phase containing 20 nm (diameter) sulfonated polymer nanoparticles improved the separation of five catecholamines.29 Although resolution in these experiments was poor, the usefulness of nanomaterials in separations and the importance of nanomaterial surface chemistry were clearly demonstrated. Recent advances in nanomaterials and various CE modes were summarized in several reviews and included: (1) separation effects of nanoparticle pseudostationary phases in CE,30,31 (2) general uses of nanomaterials in separation science,32 (3) extension and modification of MEKC mathematics to nanoparticle pseudostationary and stationary phases in CE,33 (4) exploitation of nanomaterials for electrochemical detection in CE,34 and (5) electrophoretic separations of nanopartcles.35
In this review, the importance of nanomaterial stability and surface chemistry in various modes of CE will be discussed. In particular, the characterization of nanomaterial surface chemistry for customized use in separations, properties of nanomaterials inside the capillary, methods of nanomaterial introduction, and nanomaterial surface chemistry dependent molecular interactions will be discussed.
2. Nanomaterial core classification
Research funding for the development of applications which include nanomaterials is continually increasing. Globally, the total number of nanotechnology patent applications filed in 2008 exceeded 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 among the 15 largest national patent registries.36 For the 2011 fiscal year, the United States National Nanotechnology Initiative (NNI) requested $1.8 billion for nanotechnology investments. Since 2001, the cumulative NNI investment in nanotechnology, including the 2011 request, is ∼$14 billion.37 These substantial investments are directly related to the novel size dependent chemical and physical properties of these materials at the nanoscale where catalytic,38 electrical,39 magnetic,40 mechanical,41 optical,42 and thermal43 properties can deviate from those of bulk materials.
000 among the 15 largest national patent registries.36 For the 2011 fiscal year, the United States National Nanotechnology Initiative (NNI) requested $1.8 billion for nanotechnology investments. Since 2001, the cumulative NNI investment in nanotechnology, including the 2011 request, is ∼$14 billion.37 These substantial investments are directly related to the novel size dependent chemical and physical properties of these materials at the nanoscale where catalytic,38 electrical,39 magnetic,40 mechanical,41 optical,42 and thermal43 properties can deviate from those of bulk materials.
Although nanomaterial properties are primarily dictated by composition, shape, and size; precise control of nanomaterial surface chemistry is one of the critical characteristics for successful and reproducible nanomaterial applications. The large surface area to volume ratio of nanomaterials relative to the bulk increases the overall surface energy of the nanomaterial system thereby increasing its reactivity. Surface chemistry influences the surface energy, functionality, and structural stability of nanomaterials;44 and as a result, can be used to modulate surface energy thereby dictating the function of the nanomaterial in a bulk environment.45 Additionally, because of the high surface energy of nanomaterial systems, structural changes in nanoparticles are often observed,46 and nanoparticle surface chemistry can lead to structurally stable or asymmetric nanoparticle architectures.47
Nanomaterials are ideally suited as pseudostationary and stationary phases in electrokinetic chromatography (EKC) because of their small sizes, large surface area to volume ratios vs. bulk materials, and customizable surface chemistries. Nanoparticle surface chemistry plays two important roles in these separations. First, nanoparticle surface chemistry can modulate the separation mechanism(s) (i.e. effects the electroosmotic flow (EOF) or capillary surface) and dictate the elution order of targeted molecules.24,25,48,49 For instance, Kuo et al.48 observed an increase in effective capillary surface charge as silica nanoparticle concentration (diameter, d = 60 nm) increased in pH ≈ 3 buffer.48 As nanoparticle concentration increased (∼50 to 180 nM), migration times of the analytes varied slightly as nanoparticles aggregated and/or interacted with the capillary wall. Nanomaterial instability was apparent from increased scattering intensities, unstable baselines, irreproducible separations (at elevated nanoparticle concentrations), and/or reduced detection sensitivities. Second, surface chemistry can dictate nanomaterial stability as a function of nanomaterial–nanomaterial interactions, interactions between the nanomaterial and the separation environment, and interactions between the target molecules and nanomaterial. Li et al.50 used citrate to both stabilize gold nanoparticles (d = 13 nm) and to extract indoleamines from solution prior to separations. The targeted molecules were hypothesized to interact with citrate on the nanoparticle surface via either van der Waals or electrostatic interactions. Limits of detection were improved by a factor of ∼4000 for separations using nanoparticle extraction techniques vs. controls.
In both of these previous examples, surface chemistry dictated the role of nanomaterials in the separation. As a result, all properties of the nanostructures—including core composition, shape, size, and surface chemistry—must be considered when evaluating the functionality of nanomaterials in separations. To simplify this discussion, we will classify nanostructures as containing either non-plasmonic or plasmonic cores.
2.1 Non-plasmonic nanomaterials
Non-plasmonic nanomaterials such as carbon nanotubes (CNTs), latex/polymer nanoparticles, and silica nanoparticles exhibit novel properties which are routinely exploited as pseudostationary and stationary phases in various capillary based separation techniques. For instance, the high surface area to volume ratio and conductivity of CNTs encourage their use in separations.51 CNTs are one dimensional nanostructures and can be classified as either single-walled nanotubes (SWNTs) or multi-walled nanotubes (MWNTs) which are CNTs composed of one or multiple concentric cylinder(s), respectively.Despite many promising investigations with the separation of peptides,52aniline derivatives,53 and water soluble vitamins;54 CNTs have experienced limited success in CE. First, CNTs can interfere with detectable signals from both ultraviolet (UV) and laser induced fluorescence (LIF) detectors.55 Second, raw CNTs are highly conductive56 which can lead to irreproducible currents and separations. Third, the surface of a CNT is natively hydrophobic57 which results in materials which are insoluble in water and commonly used separation buffers.
The conductivity and solubility of CNTs are influenced by surface chemistry. For instance, CNT surfaces are oxidized by concentrated acids which yield hydroxyl, carbonyl, or carboxyl head groups rendering the normally non-polar CNTs water soluble.58,59 In 2003, Wang et al.60 used this strategy to successfully incorporate SWNTs as a pseudostationary phase into EKC. Since that hallmark work, the utility of SWNTs in CE was further advanced by surfactant modified surface chemistries. Suárez et al.61 modified SWNTs (d = 0.7–1.2 nm) with sodium dodecyl sulfate (SDS) (CNT@SDS) which improved the separation resolution of three mixtures containing chlorophenols, non-steroidal anti-inflammatory drugs, or penicillin derivatives. Importantly, molecule-dependent separation selectivity was observed. When CNT@SDS was included in the run buffer, the mobility of the penicillin derivative, penicillin G, decreased vs. no additives thereby suggesting a high affinity of penicillin G to the nanomaterial surface compared to the other penicillin derivatives.
Latex nanoparticles are also used to improve the separation of pharmaceutical compounds. Recently, Palmer62 synthesized latex nanoparticles (d = 63 nm) for use as an EKC pseudostationary phase. Unlike CNTs, latex nanoparticles contain native surface chemistries that are water soluble and cores which are not conductive. In comparison to traditional MEKC separations, the average plate numbers for the separation of pharmaceutical compounds were slightly lower vs. controls (2 × 105 m−1); however, selectivity increased dramatically for the nanoparticle-based experiments. Improvements in selectivity were attributed to surface chemistry; that is, increased hydrophobic interactions between the nanoparticle surface and target molecules.
Silica nanoparticles are also widely used to improve separations. Above pH ∼2 to 3, the surface silanol groups of silica nanoparticles are negatively charged and under normal polarity conditions will force the particles to migrate towards the anode (away from the detector). Fujimoto63 exploited this native surface charge to selectively induce increased hydrogen bonding between the silica particle surface and neutral, polar organic molecules in solution. This approach was further used to improve the separation of aromatic acids64 and anti-bacterial quinolones.65
Despite pseudostationary phases offering generally higher selectivity than if no pseudostationary phase was used, possible negative impacts on resolution are often overlooked or underestimated when nanomaterials are used in CE. For instance, while not technically nanomaterials (i.e. critical dimensions are larger than 100 nm), Bächmann et al.66,67 compared separation implications of 500 nm and 1.5 µm diameter silica particles to micelles used in MEKC. The total band broadening (Htotal) was defined as follows:
| Htotal = Hl + Hm + HT+ Hep(p) + Hi |
2.2 Plasmonic nanomaterials
Plasmonic nanomaterials (i.e.gold, silver, composites, etc.) exhibit a size dependent property in which electrical and optical energy can be stored at their surfaces which is known as a localized surface plasmon resonance (LSPR).68,69 When metallic nanostructures possess critical size dimensions smaller than ∼half the wavelength of incident light, the incident electromagnetic energy can be selectively absorbed by the nanoparticle which induces an oscillating electric field localized at the nanoparticle surface.70The LSPR of noble metal nanoparticles is (1) experimentally measured using extinction spectroscopy (i.e. scattered and absorbed light);71 (2) dependent on the distance matter is from the nanoparticle surface;72 (3) theoretically predicted using Mie theory;73 and (4) dictated by nanoparticle composition, shape, size, and local environment surrounding the core nanomaterial.73–75 Previously, LSPR spectroscopy was used to investigate a wide variety of chemical phenomena including biosensors for disease detection,76 cancer research,77,78 single molecule detection,79 and surface enhanced Raman scattering (SERS).80
The influence of surface chemistry on the local environment and LSPR of gold nanoparticles (d ≈ 13 nm) is demonstrated in Fig. 2. Gold nanoparticles functionalized with citrate, thioctic acid, 11-mercaptoundecanoic acid, and 6-mercaptohexanoic acid display unique extinction spectra with characteristic extinction maximum wavelengths (λmax) (Fig. 2). While all four nanoparticle samples are statistically identical in terms of core sizes, the citrate stabilized gold nanoparticles contain the lowest molecular density of electrostatically bound surface molecules, and as a result, the extinction maximum of the sample is blue-shifted from the three covalently bound functional groups.
 | ||
| Fig. 2 Gold nanoparticles functionalized with (1) citrate; (2) thioctic acid; (3) 6-mercaptohexanoic acid; and (4) 11-mercaptoundecanoic acid. (Top panel) Reaction scheme for functionalization (not to scale). (Bottom panel) Normalized extinction spectra in aqueous solutions: (1) λmax = 519.7 nm; (2) λmax = 521.1 nm; (3) λmax = 527.7 nm; and (4) λmax = 529.5 nm. | ||
The nanostructures functionalized with carboxylic acid terminated self-assembled monolayers (SAMs) exhibit surface chemistry dependent trends as well. As shown in Fig. 2B, the extinction maximum wavelength red shifts as molecular packing density and/or alkanethiol chain length increases. For example, it is well-established that thioctic acid binds to the gold nanoparticle surface via a disulfide ring,81 whereas 11-mercaptoundecanoic acid and 6-mercaptohexanoic acid are straight chain alkanethiols. The larger binding footprint of thioctic acid lowers its packing density vs. the other two SAMs.82 Furthermore, because the LSPR wavelength increases as alkanethiol chain length increases,7411-mercaptoundecanoic acid shows a lower energy extinction maximum than 6-mercaptohexanoic acid. These surface chemistry-dependent properties can serve as an important monitoring parameter for understanding nanoparticle function and stability during electrically driven capillary based separations.
3. Bulk nanomaterial characterization
Nanomaterials exhibit surface chemistry dependent properties which provide numerous advantages for their use in separation science. Nanomaterial stability and utility in CE are dictated by their core composition and surface chemistry. As a result, thorough characterization of these properties as well as an evaluation of their shape, size, and reactivity are needed to establish a mechanistic understanding of how nanomaterials impact separations. If this information is not evaluated, the ability to use these materials reproducibly and to predict structure–function behaviour is difficult/impossible.3.1 Core vs. surface classification
Classification of nanomaterial pseudostationary and stationary phases in various capillary based separation techniques is summarized in Fig. 3. The nanomaterials are divided into two categories: (1) nanomaterial cores modified by a capping agent (i.e. chemically distinct surface chemistry) that differs in composition from the core: “core ≠ surface” and (2) nanomaterial cores modified by native surface functionality of the nanomaterial: “core = surface” (i.e. core and surface are chemically identical but contain distinct atomic coordination numbers or a native oxide surface layer). | ||
| Fig. 3 Classification chart of nanomaterial use in capillary electrophoresis. Nanomaterials are divided into two general categories core = surface and core ≠ surface. | ||
The chemical diversity of nanomaterials is complex as both core and surface chemistry composition dictate material properties and function (Fig. 3). First, surface chemistry homogeneity impacts the stability of a nanostructure. For instance, charge density is often more homogeneous if capping agents are used vs. native functionalization (core = surface);83,84 however, non-specific molecular binding to “core = surface” structures is inherently less controllable than if a capping agent is used.
Furthermore, in “core ≠ surface” nanostructures, molecules can form localized distributions with distinct molecular orientations and packing densities on a single nanoparticle which can vary from nanoparticle to nanoparticle.85 As a result, nanomaterial interactions can differ slightly from nanoparticle to nanoparticle thereby yielding an overall more heterogeneous nanomaterial phase than if the nanoparticle is stabilized by its native surface functionality (core = surface).
Finally, because initial surface functionalization is often used for both core stabilization and subsequent conjugation,86–88 both head and tail groups on a surface ligand must be chosen for successful integration into applications. Because capping agents (core ≠ surface) offer more options for chemical flexibility and materials stability than native surface chemistries, this nanoparticle design approach is more often employed in capillary based separations. Both platforms (core = surface and core ≠ surface), however, yield nanoparticle surface chemistries that can be further exploited for their electrostatic, covalent, and/or reversible functionalities.
3.2 Nanomaterial stability
Surface chemistry is particularly important in preventing uncontrolled nanomaterial aggregation during use in all applications including in separations. Because background electrolyte solutions are used in a wide range of ionic strengths and pHs, the stability of potential nanomaterial additives is dictated by the capping agent pKa and composition. These properties govern nanoparticle stability which is dictated by electrostatic forces,89hydrogen bonding,90,91 and/or van der Waals interactions.92For example, Preston et al.93 successfully demonstrated the reversible agglomeration of gold nanoparticles (d = 11 nm) functionalized with various SAMs (6-mercaptohexanoic acid, 12-mercaptododecanoic acid, and N-(2-(S)-methylacetic acid)-6-mercaptohexamide) (Fig. 4). Using Derjaguin, Landau, Verwey and Overbeck (DLVO) theory,94 the kinetic stability of these carboxylic acid functionalized gold nanoparticles was dictated by both electrostatic and van der Waals interactions.93 Once initial aggregation occurred via van der Waals interactions, hydrogen bonding was hypothesized to stabilize the surface interactions and consequently, the large aggregates.
 | ||
| Fig. 4 Model of nanoparticle–nanoparticle surface chemistry interactions. (A) The radius, r, capping agent coating thickness, δ, and inter-particle surface chemistry distance, h, are represented. (B) Chemical structures of capping agents used to coat gold nanoparticles. (1) 6-mercaptohexanoic acid, (2) 12-mercaptododecanoic acid, and (3) N-(2-(S)-methylacetic acid)-6-mercaptohexamide (reprinted with permission from ref. 93, Copyright 2009, American Chemical Society). | ||
3.3 Nanomaterial concentration
While numerous techniques are routinely used to characterize noble metal nanoparticles, one of the most widely used for estimating plasmonic nanoparticle concentration exploits the LSPR. For instance, the concentration of gold nanoparticles can be assessed using a standard estimation model95 where LSPR spectroscopy and transmission electron microscopy (TEM) data are utilized. In addition to estimating nanoparticle concentration, LSPR spectral changes for noble metal nanoparticles45,96 can indicate nanoparticle stability and surface chemistry properties (Fig. 2).Conversely, in cases where LSPR data are neither available nor exploited, nanomaterial concentration can be reported as a weight/weight (w/w) or weight/volume (w/v) ratio. These methods depend on measuring and suspending a known initial dry weight of a nanomaterial in solution. If an average nanoparticle size is reported, nanoparticle concentration can be estimated. Alternatively, nanomaterial concentration can be calculated from the amount of starting reagents used. The morphology of the nanomaterials is then used to estimate concentration.
3.4 Nanomaterial surface charge
Nanomaterial stability and their potential usefulness as an additive in capillary-based separation techniques are directed by surface chemistry and charge. ζ-Potential, a technique that measures electric potential differences between the slipping planes of a stationary fluid layer on a nanoparticle surface,97 can be used to quantify surface charge in a fixed buffer composition, concentration, ionic strength, and pH environment. In general, a “high” ζ-potential (25 mV or −25 mV) indicates stable nanostructures. As the ζ-potential approaches “0” or the point of zero charge (via pH changes),98,99 the attraction between nanomaterials exceeds the repulsive forces between the structures resulting in agglomeration.100 As a result, this solution-dependent surface charge measurement is useful in predicting the stability and function of nanomaterials in various environments.3.5 Surface functionality
While both LSPR and ζ-potential data yield important information regarding the composition, concentration, and effective charge of nanomaterials, more precise information regarding molecule orientation must be garnered using other techniques such as X-ray photoelectron spectroscopy (XPS)101,102 and NMR.103,104Traditionally used as an organic chemistry characterization tool, NMR can provide detailed information regarding the interactions between105,106 as well as orientation and composition of104,107 SAMs on nanomaterials. For instance, Schmitt et al. used solid state 13C NMR to investigate the effects of SAM terminal groups on ∼3 nm gold nanoparticles.108 The conformational ordering of 8-mercapto-1-octanoic acid and 16-hydroxyhexadecanethiol was dictated by the degree of (1) van der Waals interactions between the alkanethiol chains and (2) hydrogen bonding between terminal groups of neighbouring molecules on individual nanoparticles. Carboxylic acid terminated SAMs exhibited the most conformational order followed by hydroxylated and methyl terminated functionalities.
4. Incorporating nanomaterials in CE
Manipulation of nanomaterial pseudostationary and stationary phase properties (i.e. core composition, shape, size, and surface chemistry) inherently impacts their function in capillary based separation techniques. Once the nanomaterial core and surface compositions are selected and evaluated, the method in which the nanomaterials are introduced into a capillary is the next critical parameter to optimize both stable and reproducible applications. Three methods are typically used to introduce nanomaterials as pseudostationary and stationary phases into a capillary: continuous full filling,109–112 partial filling,61,110,113 and capillary coating.24,25,109,112,114–116The first deliberate use of nanomaterials29 in EKC implemented continuous full filling methods where the nanomaterials were included in the background electrolyte solution. Target molecules interacted with the nanomaterials throughout the separation. Partial filling techniques, where discrete nanomaterial plugs are injected into a capillary prior to the sample matrix, were successfully used to generate discontinuous nanomaterial pseudostationary phases. Because target molecules possessed greater mobilities than the nanoparticle pseudostationary phase, the molecules eluted through the nanomaterial pseudostationary phase thereby influencing separation selectivity.61,110,113 Regardless of whether the nanomaterials are injected into the capillary as a discrete plug or buffer additive, the method of injection (hydrodynamic or electrokinetic) can bias the nanomaterial concentration and drastically impact the nanomaterial function in a separation. Finally, capillary coating methods utilize nanomaterials through capillary attachment via covalent and/or electrostatic interactions.24,25 In these cases, the nanomaterial is modeled as a stationary phase.
In all of these nanomaterial integration techniques, optimization of separation conditions is required for reproducible separation results. Control (i.e. lack of control or understanding) of nanomaterial concentration can adversely impact separations by (1) causing instability in the run buffer, (2) interfering with detection by generating an inconsistent background signal, (3) inducing conductivity differences within the capillary, and (4) increasing backpressure within the capillary.
4.1 Separating nanoparticles based on size
Surface chemistry is an important tool in the prevention of uncontrolled nanoparticle aggregation in commonly used CE buffers, in the presence of target molecules, on the capillary wall, and/or with other nanomaterials during a separation. Furthermore, surface chemistry is also used to separate nanomaterials based on size.117–125For example, Liu126 used the size dependent mobility of citrate functionalized gold (Au@citrate) nanoparticles to determine an unknown nanoparticle size with similar core and surface compositions. To realize this, a calibration curve was generated from the migration time of a Au@citrate nanoparticle plug as a function of nanoparticle diameter (d = 5–40 nm). Next, an unknown Au@citrate nanoparticle sample was evaluated and their diameter was predicted from the calibration curve. Finally, the results were verified using scanning electron microscopy (correlation coefficient = 0.995).
Migration times of a nanoparticle plug injected in a capillary can also be used to evaluate and predict surface chemistry. For example, 40 nm nanoparticles functionalized with varying ratios of surface ligands 3-(aminopropyl)triethoxysilane (APTES) and 2-[methyoxy(polyethyleneoxy)propyl]-trimethoxysilane (PEPS) were injected into a capillary as a discrete plug during a reverse phase separation.127 As shown in Fig. 5, the nanomaterials were positively charged from the protonated surface amine groups (NH3+) which systematically increased as the surface ligand ratio increased. Consequently, nanoparticle electrophoretic mobility increased and was attributed to an increased positive surface charge density on the nanoparticles. Furthermore, as the surface ligand ratio increased, ligand packing density and nanoparticle mobility eventually saturated, and analyte resolution decreased. These results suggest that surface chemistry is important in predicting and understanding nanomaterial mobility and stability during a separation.
 | ||
| Fig. 5 Electrophoretic mobility (µep) of amino/PEG-functionalized core/shell particle types as a function of eight different APTES/PEPS molar ratios (0.25, 0.35, 0.5, 0.75, 1, 1.25, 1.5 and 1.75) (reprinted with permission from ref. 127, Copyright 2009, Wiley). | ||
4.2 Monitoring inter-nanoparticle interactions
In capillary based separations, nanomaterials can interact with target molecules, the capillary wall, and/or other nanomaterials during a separation. Assessing nanoparticle stability during a separation is difficult as most nanomaterials do not exhibit aggregation-dependent properties that can be easily monitored in a capillary. One noteworthy exception is the LSPR spectra of noble metal nanoparticles.Recently, Ivanov et al.113 demonstrated that LSPR spectroscopy can be integrated with capillary zone electrophoresis to monitor nanoparticle–nanoparticle interactions. A discrete plug of gold nanoparticles (d = 13 nm) with covalently bound amine, covalently bound carboxyl, or electrostatically stabilized citrate functionalities was injected into a capillary. The initially discrete nanoparticle plug migrated through the capillary, and extinction intensities at 520 (I520) and 600 (I600) nm were monitored. The ratio of these data coupled with peak migration times and widths indicated the degree of nanoparticle agglomeration. Stable nanoparticles were characterized by a I520:I600 ratio of ∼3.5 to 4, a value consistent with stable nanoparticles outside the capillary. Upon agglomeration, this ratio decreased and the peak width and migration times in the electropherograms increased systematically until uncontrolled aggregation occurred.
These data revealed surface chemistry-dependent trends. As a result, a critical nanoparticle concentration (CNC) was developed to quantify nanoparticle stability (for a fixed size, core composition, and surface chemistry) under specific separation conditions. The CNC, a parameter similar to the critical micelle concentration in MEKC, was defined as the lowest nanoparticle concentration that induced uncontrolled nanoparticle agglomeration in the capillary. This value ranged from 2.3 nM to 1.8 nM for aminated and carboxylated nanostructures, respectively.
The degree of inter-nanoparticle interactions can also be monitored indirectly using chemiluminescence.128 Specifically, the catalytic activity of Au@citrate nanoparticles as a function of diameter (d = 16–99 nm and included in the run buffer) was monitored viachemiluminescence detection of luminol–H2O2. The greatest catalytic effect was monitored for 38 nm Au@citrate nanoparticles and occurred in a nanoparticle concentration-dependent manner (saturated at 150 µM HAuCl4). Above this concentration, the chemiluminescence intensity decreased. While it was not stated, Au@citrate nanoparticles are typically unstable at these high concentrations and were likely above their CNC. As a result, the loss of catalytic activity could be a result of nanoparticle agglomeration or instability.
Non-plasmonic nanomaterials also exhibit indirect evidence of inter-nanoparticle interactions in EKC. Wang65 demonstrated that silica nanoparticles (d = 30 nm) increased EOF (pH = 9) in a concentration dependent manner from ∼2 to ∼5 µg mL−1. No systematic trends in EOF, however, were observed for silica nanoparticles above ∼5 µg mL−1. We postulate that this concentration represents a CNC for these nanomaterials and that nanoparticle aggregation was likely occurring at these relatively high concentrations.
In another study, Kuo et al. observed an increase in effective capillary surface charge with increasing concentrations of silica nanoparticles (d = 60 nm, normal polarity, pH < 4).48 As nanoparticle concentration approached ∼180 nM, the nanomaterials aggregated. Nanomaterial instability was apparent from increased scattering intensities, unstable electropherogram baselines, irreproducible separations, and reduced detection sensitivity (increased baseline noise).
4.3 Modulating nanomaterial-analyte interactions
Improving the separation of target molecules using nanomaterial pseudostationary and stationary phases requires consideration of both the targeted analyte(s) and nanoparticle functionalities. For example, electrostatic interactions between dopamine and negatively charged carboxylated (Au@COO−) vs. positively charged aminated (Au@NH3+) gold nanoparticles (d = 13 nm) induced opposite effects on the apparent mobility of dopamine (pH = 9.3 run buffer).113 In Fig. 6, a discrete plug of either Au@COO− or Au@NH3+ nanoparticles was injected into a capillary before a dopamine-containing sample matrix. In both cases, migration time differences increased as nanoparticle concentration increased. For Au@NH3+ nanoparticles, however, dopamine experienced electrostatic repulsive forces which led to an increase in its mobility. Conversely the mobility of dopamine decreased when a Au@COO−nanoparticle pseudostationary phase was used. These concentration and surface chemistry-dependent nanoparticle effects were systematically observed for stable nanoparticles, that is, at concentrations less than their CNC. | ||
| Fig. 6 Comparison of dopamine mobility versusnanoparticle concentration for (A) Au@COO− and (B) Au@NH3+ nanoparticles (reprinted with permission from ref. 113, Copyright 2009, American Chemical Society). | ||
Recently, Nilsson et al.129 demonstrated that lipid based crystalline nanoparticles130 (d = 70 nm) can improve the separation of a protein mixture. Ordinarily, proteins exhibit high affinities for co-polymer capillary walls which reduce separation efficiency and reproducibility. When lipid nanoparticles were included, the nanostructures exhibited a higher affinity for the capillary wall vs. the proteins thereby suppressing protein adsorption on the capillary wall. It should be noted that nanoparticle concentration was high (2% w/w), and nanoparticle–protein interactions were minimized thereby facilitating separations with ∼4 × 105 plates m−1.
Selective analyte partitioning between mobile (buffer) and stationary (nanoparticle surface chemistry) phases can be used to improve separation efficiency. EKC separations which include nanomaterials that are designed to exploit partitioning typically do not quantify separation parameters such as band broadening or partition coefficients. Nonetheless, partition coefficients attained outside EKC platforms can be used to estimate what is occurring inside a capillary.
For instance, Lucarini et al.131,132 characterized these interactions using electron spin resonance (ESR) spectroscopy. The partition coefficients (Keq) for the hydrophobic free-radical target molecule p-pentylbenzyl hydroxyalkyl nitroxide between an aqueous solvent and the hydrophobic thiolated triethylene glycol SAM on gold nanoparticle surfaces (d = 1.6–5.3 nm) were determined. A ratio of the free radical (nitroxide) bound to the monolayer to that free in solution was plotted against the total SAM molecule concentration (Fig. 7). The slope from these data yielded a Keq and revealed that partitioning increased with decreasing nanoparticle diameter which reduced the energy required for molecules to partition into the SAM.
![Determination of the equilibrium constant (Keq) of lipophilic nitroxides partitioned into a gold nanoparticle SAM. Keq is determined from the ratio of lipophilic nitroxides partitioned into the gold nanoparticle monolayer (lipophilic nitroxidesSAM) and free species in solution (lipophilic nitroxidesH2O) to the total gold nanoparticle monolayer concentration [SAM]. Gold nanoparticle diameters = ● 1.6 nm; ▼ 3.4 nm; and ■ 5.3 nm (reprinted with permission from ref. 132, Copyright 2005, American Chemical Society).](/image/article/2011/AN/c0an00458h/c0an00458h-f7.gif) | ||
| Fig. 7 Determination of the equilibrium constant (Keq) of lipophilic nitroxides partitioned into a gold nanoparticle SAM. Keq is determined from the ratio of lipophilic nitroxides partitioned into the gold nanoparticle monolayer (lipophilic nitroxidesSAM) and free species in solution (lipophilic nitroxidesH2O) to the total gold nanoparticle monolayer concentration [SAM]. Gold nanoparticle diameters = ● 1.6 nm; ▼ 3.4 nm; and ■ 5.3 nm (reprinted with permission from ref. 132, Copyright 2005, American Chemical Society). | ||
4.4 Influencing analytes with nanomaterials in CE
In all electrically driven (capillary) separations, capillary coatings (i.e. capillary surface chemistry) dictate the mode of separation (normal or reverse phase) as well as the magnitude and direction of EOF.133,134 When nanomaterial pseudostationary and/or stationary phases are used in EKC or CEC (Fig. 1), nanoparticle surface chemistry can provide an additional advantage of reducing non-specific adsorption of analytes onto capillary walls.Yu et al.24 studied the effects of acidic and basic proteins in a capillary that contained gold nanoparticles (d = 6 nm) functionalized with two layers: initially with didodecyldimethylammonium bromide (Au@DDAB) and additionally modified with poly(ethylene oxide) (Au@DDAB + PEO nanoparticles). Typically, DDAB is used (without nanoparticles) as a capillary coating to prevent non-specific adsorption of biomolecules to capillary walls.135,136 In Fig. 8, Au@DDAB and PEO nanoparticles were utilized in a separation buffer and capillary coating to interact with target molecules via both hydrophobic and covalent interactions (through cysteine and lysine residues). Under acidic separation conditions (pH < 4), proteins with acidic and basic isoelectric points were poorly resolved in DDAB coated capillaries (no nanoparticles). When Au@DDAB and PEO nanoparticles were included, a PEO concentration-dependent improvement in plate number was observed up to ∼6.24 × 105 plates m−1 for the same molecules.
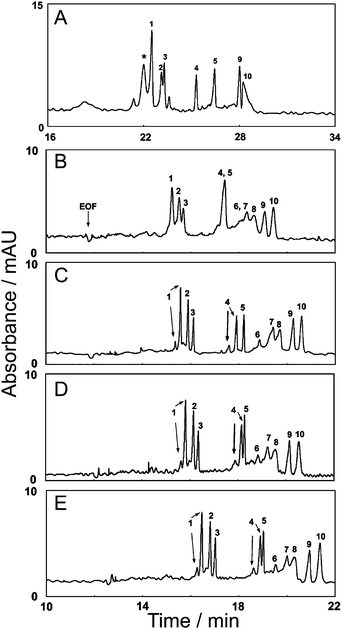 | ||
| Fig. 8 Evaluation of the separation of acidic and basic proteins in a DDAB coated capillary using 25 mM phosphate buffer. Separation A contained no Au@DDAB nanoparticles whereas separations B–E contained ∼4.5 nM Au@DDAM nanoparticles modified with (B) 0.01, (C) 0.05, (D) 0.07, and (E) 0.10% PEO. Peak identification: (1) α-chymotrypsinogen, (2) ribonuclease A, (3) trypsinogen, (4) cytochrome c, (5) lysozyme, (6) bovine serum albumin, (7) carbonic anhydrase, (8) ovalbumin, (9) myoglobin, and (10) α-lactalbumin (reprinted with permission from ref. 24, Copyright 2006, American Chemical Society). | ||
5. Conclusions and future outlook
Nanomaterials are widely used as pseudostationary and stationary phases in electrically driven capillary separations. Nanomaterial advantages are numerous and include small/tuneable sizes, core composition variations, flexible injection/introduction methods in separation techniques, and diverse surface chemistry options. Nanomaterials, however, exhibit inherently large surface energies which can change upon aggregation and/or surface chemistry modification, and as a result, yield unpredictable function in separations. Furthermore, nanomaterials can adversely impact separations by changing buffer conductivity, viscosity, and pH which requires a careful balance in nanoparticle stability and separation optimization. Finally, nanomaterials can complicate detection by varying background signals.A unified mechanism which predicts the structure–function relationship of a nanomaterial phase to separation effects is complex, dynamic, and not often thoroughly understood. To accomplish a better mechanistic understanding of these interactions, more rigorous evaluations of nanoparticle core and surface properties must be included in separation studies. Once this is done, a more unified understanding of how nanomaterials impact important separation parameters such as efficiency, resolution, and selectivity will be more easily developed.
Without this thorough evaluation of nanoparticle properties, however, exciting and promising separation studies are being performed in many laboratory settings. To date, most of these studies probed the interactions between nanomaterials with capillary surfaces, target analytes, and other nanoparticles. Additional evaluation of inter-nanoparticle interactions will likely improve these separation results thereby providing a better mechanistic understanding of the nanomaterial function. Clearly, an interdisciplinary approach which couples nanomaterial characterization methods (for both core and surface properties) used outside the capillary to data collected inside the capillary will be required to achieve this level of understanding.
The use of nanomaterial pseudostationary and stationary phases in separations will continue to expand because of resulting improvements in both separation selectivity and detection. Regardless of the core nanomaterial, injection/introduction approach, or detection method used; nanomaterial surface chemistry will continue to drive their mechanisms in these separations.
Acknowledgements
This work was funded by the Roy J. Carver Charitable Trust and NIH-NCRR 1UL1RR024979, 1KL2RR024980 and 1TL1RR024981—University of Iowa Clinical and Translational Science Program. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Institutes of Health.Notes and references
- A. Tiselius, Trans. Faraday Soc., 1937, 33, 524 RSC.
- S. Hjerten, Chromatogr. Rev., 1967, 9, 122–219 CrossRef CAS.
- J. W. Jorgenson and K. D. Lukacs, Anal. Chem., 1981, 53, 1298–1302 CrossRef.
- T. Tsuda, K. Nomura and G. Nakagawa, J. Chromatogr., A, 1983, 264, 385–392 CrossRef CAS.
- M. Huang, Y. Kuo, C. Huang and H. Chang, Anal. Chem., 2004, 76, 192–196 CrossRef CAS.
- B. K. Clark, T. Vo-Dinh and M. J. Sepaniak, Anal. Chem., 1995, 67, 680–683 CrossRef CAS.
- D. M. Goodall, S. J. Williams and D. K. Lloyd, TrAC, Trends Anal. Chem., 1991, 10, 272–279 CrossRef CAS.
- D. J. Rose and J. W. Jorgenson, Anal. Chem., 1988, 60, 642–648 CrossRef CAS.
- B. L. Karger and F. Foret, in Capillary Electrophoresis: Introduction and Assessment, Marcel Dekker, New York, New York, USA, 1993 Search PubMed.
- K. Altria, J. Chromatogr., A, 1999, 856, 443 CrossRef CAS.
- S. F. Y. Li, in Capillary Electrophoresis: Principles, Practice and Applications, Elsevier, Amsterdam, 1992 Search PubMed.
- S. Terabe, K. Otsuka, K. Ichikawa, A. Tsuchiya and T. Ando, Anal. Chem., 1984, 56, 111–113 CrossRef CAS.
- S. Terabe, K. Otsuka and T. Ando, Anal. Chem., 1985, 57, 834–841 CrossRef CAS.
- M. J. Sepaniak, D. E. Burton and M. P. Maskarinec, ACS Symp. Ser., 1987, 342, 142–151 CAS.
- N. Hiroyuki, J. Chromatogr., A, 1991, 553, 503–516 CrossRef.
- J. Vindevogel and P. Sandra, Anal. Chem., 1991, 63, 1530–1536 CrossRef CAS.
- T. Yashima, A. Tsuchiya, O. Morita and S. Terabe, Anal. Chem., 1992, 64, 2981–2984 CrossRef CAS.
- R. M. Seifar, J. C. Kraak and W. T. Kok, Anal. Chem., 1997, 69, 2772–2778 CrossRef CAS.
- J. W. Jorgenson and K. D. Lukacs, J. Chromatogr., A, 1981, 218, 209–216 CrossRef CAS.
- A. S. Cohen and B. L. Karger, J. Chromatogr., A, 1987, 397, 409–417 CrossRef CAS.
- C. Huhn, R. Ramautar, M. Wuhrer and G. Somsen, Anal. Bioanal. Chem., 2010, 396, 297–314 CrossRef CAS.
- U. Pyell, in Fundementals of Capillary Electrophoresis, Marcel Dekker, New York, 2001 Search PubMed.
- E. Wen, R. Asiaie and C. Horváth, J. Chromatogr., A, 1999, 855, 349–366 CrossRef CAS.
- C. Yu, C. Su and W. Tseng, Anal. Chem., 2006, 78, 8004–8010 CrossRef CAS.
- Y. Hsieh, Electrophoresis, 2005, 26, 4089–4097 CrossRef CAS.
- R. Freitag, J. Chromatogr., A, 2004, 1033, 267–273 CrossRef CAS.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- R. A. Wallingford and A. G. Ewing, Adv. Chromatogr., 1989, 29, 1–76 CAS.
- C. Nilsson and S. Nilsson, Electrophoresis, 2006, 27, 76–83 CrossRef.
- C. Nilsson, S. Birnbaum and S. Nilsson, J. Chromatogr., A, 2007, 1168, 212–224 CrossRef CAS.
- E. Guihen and J. D. Glennon, Anal. Lett., 2003, 36, 3309–3336 CrossRef CAS.
- B. Gottlicher and K. Bachmann, J. Chromatogr., A, 1997, 780, 63–73 CrossRef CAS.
- M. Pumera and A. Escarpa, Electrophoresis, 2009, 30, 3315–3323 CrossRef CAS.
- N. Surugau and P. L. Urban, J. Sep. Sci., 2009, 32, 1889–1906 CrossRef CAS.
- Y. Dang, Y. Zhang, L. Fan, H. Chen and M. Roco, J. Nanopart. Res., 12, 687 Search PubMed.
- 21st Century Nanotechnology Research and Development Act, 2003.
- M. Grass, J. Phys. Chem. C, 2009, 113, 8616–8623 CrossRef CAS.
- S. B. Yang, J. Phys. Chem. C, 2010, 114, 9296–9300 CrossRef CAS.
- H. Hori, T. Yamamato and T. Iwamoto, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 174411 CrossRef.
- S. Smart, J. Appl. Polym. Sci., 2010, 117, 24–32 CAS.
- C. L. Nehl and J. H. Hafner, J. Mater. Chem., 2008, 18, 2415–2419 RSC.
- P. Buffat, Phys. Rev. A: At., Mol., Opt. Phys., 1976, 13, 2287–2298 CrossRef CAS.
- L. Srisombat, J.-S. Park, S. Zhang and T. R. Lee, Langmuir, 2008, 24, 7750–7754 CrossRef CAS.
- C. S. Weisbecker, M. V. Merritt and G. M. Whitesides, Langmuir, 1996, 12, 3763–3772 CrossRef CAS.
- S. Lee, Y. Jun, S. Cho and J. Cheon, J. Am. Chem. Soc., 2002, 124, 11244–11245 CrossRef CAS.
- C. J. Murphy, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS.
- I. Kuo, Y. Huang and H. Chang, Electrophoresis, 2005, 26, 2643–2651 CrossRef CAS.
- W. Wang, L. Zhao, F. Zhou, J. Zhu and J. Zhang, Talanta, 2007, 73, 534–539 CrossRef CAS.
- M. Li, W. Tseng and T. Cheng, J. Chromatogr., A, 2009, 1216, 6451–6458 CrossRef CAS.
- L. Agüí, P. Yáñez-Sedeño and J. M. Pingarrón, Anal. Chim. Acta, 2008, 622, 11–47 CrossRef.
- Y. Li, Y. Chen, R. Xiang, D. Ciuparu, L. D. Pfefferle, C. Horvãth and J. A. Wilkins, Anal. Chem., 2005, 77, 1398–1406 CrossRef CAS.
- J. H. T. Luong, P. Bouvrette, Y. Liu, D. Yang and E. Sacher, J. Chromatogr., A, 2005, 1074, 187–194 CrossRef CAS.
- J. M. Jiménez-Soto, Y. Moliner-Martínez, S. Cárdenas and M. Valcárcel, Electrophoresis, 2010, 31, 1–8 CrossRef.
- Y. Xu and S. F. Y. Li, Electrophoresis, 2006, 27, 4025–4028 CrossRef CAS.
- W. Zhou, X. Bai, E. Wang and S. Xie, Adv. Mater., 2009, 21, 4565–4583 CrossRef CAS.
- J. L. Bahr, E. T. Mickelson, M. J. Bronikowski, R. E. Smalley and J. M. Tour, Chem. Commun., 2001, 193–194 RSC.
- W. Zhao, C. Song and P. E. Pehrsson, J. Am. Chem. Soc., 2002, 124, 12418–12419 CrossRef CAS.
- J. Chen, A. M. Rao, S. Lyuksyutov, M. E. Itkis, M. A. Hamon, H. Hu, R. W. Cohn, P. C. Eklund, D. T. Colbert, R. E. Smalley and R. C. Haddon, J. Phys. Chem. B, 2001, 105, 2525–2528 CrossRef CAS.
- Z. Wang, G. Luo, J. Chen, S. Xiao and Y. Wang, Electrophoresis, 2003, 24, 4181–4188 CrossRef CAS.
- B. Suárez, B. M. Simonet, S. Cárdenas and M. Valcárcel, Electrophoresis, 2007, 28, 1714–1722 CrossRef CAS.
- C. Palmer, Anal. Chem., 2010, 82, 4046–4054 CrossRef CAS.
- C. Fujimoto, J. High Resolut. Chromatogr., 1997, 20, 400–402 CrossRef CAS.
- D. Liu, Electrophoresis, 2008, 29, 863–870 CrossRef CAS.
- Y. Wang, Talanta, 2009, 77, 1667 CrossRef CAS.
- K. Bächmann, B. Göttlicher, I. Haag, K.-Y. Han, W. Hensel and A. Mainka, J. Chromatogr., A, 1994, 688, 283–292 CrossRef CAS.
- K. Bächmann and B. Göttlicher, Chromatographia, 1997, 45, 249–254.
- D. J. Campbell and Y. Xia, J. Chem. Educ., 2007, 84, 91–96 CrossRef CAS.
- A. J. Haes, S. Zou, J. Zhao, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2006, 128, 10905–10914 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- M. D. Malinsky, K. L. Kelly, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2001, 123, 1471–1482 CrossRef CAS.
- M. S. Golden, A. C. Bjonnes and R. M. Georgiadis, J. Phys. Chem. C, 2010, 114, 8837–8843 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- A. J. Haes, S. Zou, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. B, 2004, 108, 6961–6968 CrossRef CAS.
- Y. Xia and N. J. Halas, MRS Bull., 2005, 30, 338–346 CAS.
- A. J. Haes, W. P. Hall, L. Chang, W. L. Klein and R. P. Van Duyne, Nano Lett., 2004, 4, 1029–1034 CrossRef CAS.
- R. Sinha, G. J. Kim, S. Nie and D. M. Shin, Mol. Cancer Ther., 2006, 5, 1909–1917 CrossRef CAS.
- A. G. Cuenca, H. Jiang, S. N. Hochwald, M. Delano, W. G. Cance and S. R. Grobmyer, Cancer, 2006, 107, 459–466 CrossRef CAS.
- D. J. Campbell, B. R. Herr, J. C. Hulteen, R. P. Van Duyne and C. A. Mirkin, J. Am. Chem. Soc., 1996, 118, 10211–10219 CrossRef CAS.
- A. Campion and P. Kambhampati, Chem. Soc. Rev., 1998, 27, 241–250 RSC.
- M. Rooth and A. M. Shaw, J. Phys. Chem. C, 2007, 111, 15363–15369 CrossRef CAS.
- V. Subramaniam, M. R. Ivanov, A. A. Volkert, A. M. Jones and A. J. Haes, 2010, submitted.
- R. L. Jones, N. C. Pearsall and J. D. Batteas, J. Phys. Chem. C, 2009, 113, 4507–4514 CrossRef CAS.
- G. A. DeVries, F. R. Talley, R. P. Carney and F. Stellacci, Adv. Mater., 2008, 20, 4243–4247 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 1999, 33, 27–36.
- M. J. Hostetler, A. C. Templeton and R. W. Murray, Langmuir, 1999, 15, 3782–3789 CrossRef CAS.
- J. M. Abad, S. F. L. Mertens, M. Pita, V. M. Fernandez and D. J. Schiffrin, J. Am. Chem. Soc., 2005, 127, 5689–5694 CrossRef CAS.
- J. Lyklema, in Fundamentals of Interface and Colloidal Science: Volume IV Particulate Colloids, Elsevier, Amsterdam, 2005 Search PubMed.
- S. Si and T. K. Mandal, Langmuir, 2006, 23, 190–195.
- A. Yang and C. Weng, J. Phys. Chem. C, 2010, 114, 8697–8709 CrossRef CAS.
- Y. Shiraishi, D. Arakawa and N. Toshima, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 8, 377–383 Search PubMed.
- T. C. Preston, M. Nuruzzaman, N. D. Jones and S. Mittler, J. Phys. Chem. C, 2009, 113, 14236–14244 CrossRef CAS.
- E. J. Verwey, in Theory of the Stability of Lyophilic Colloids, Elsevier, Amsterdam, 1948 Search PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS.
- K. S. Mayya, V. Patil and M. Sastry, Langmuir, 1997, 13, 3944–3947 CrossRef CAS.
- P. C. Hiemenz and R. Rajagopalan, in Principles of Colloid and Surface Chemistry, Marcel Dekkar, New York, 1997 Search PubMed.
- G. A. Parks, Chem. Rev., 1965, 65, 177–198 CrossRef CAS.
- M. Kosmulski and C. Saneluta, J. Colloid Interface Sci., 2004, 280, 544–545 CrossRef CAS.
- R. A. Alvarez-Puebla, E. Arceo, P. J. G. Goulet, J. J. Garrido and R. F. Aroca, J. Phys. Chem. B, 2005, 109, 3787–3792 CrossRef CAS.
- J. N. Park, A. J. Forman, W. Tang, J. H. Cheng, Y. S. Hu, H. F. Lin and E. W. McFarland, Small, 2008, 4, 1694–1697 CrossRef CAS.
- S. Zhang, G. Leem, L. Srisombat and T. R. Lee, J. Am. Chem. Soc., 2007, 130, 113–120.
- D. V. Leff, L. Brandt and J. R. Heath, Langmuir, 1996, 12, 4723–4730 CrossRef CAS.
- H. Y. Zhou, F. F. Du, X. Li, B. Zhang, W. Li and B. Yan, J. Phys. Chem. C, 2008, 112, 19360–19366 CrossRef CAS.
- A. Badia, L. Demers, L. Dickinson, F. G. Morin, R. B. Lennox and L. Reven, J. Am. Chem. Soc., 1997, 119, 11104–11105 CrossRef CAS.
- O. Kohlmann, W. E. Steinmetz, X. A. Mao, W. P. Wuelfing, A. C. Templeton, R. W. Murray and C. S. Johnson, J. Phys. Chem. B, 2001, 105, 8801–8809 CrossRef CAS.
- R. H. Terrill, T. A. Postlethwaite, C.-H. Chen, C. Poon, A. Terzis, A. Chen, J. E. Hutchison, M. R. Clark and G. Wignall, J. Am. Chem. Soc., 1995, 117, 12537–12548 CrossRef CAS.
- H. Schmitt, A. Badia, L. Dickinson, L. Reven and R. B. Lennox, Adv. Mater., 1998, 10, 475–480 CrossRef CAS.
- B. Neiman, E. Grushka and O. Lev, Anal. Chem., 2001, 73, 5220–5227 CrossRef CAS.
- P. Viberg, M. Jornten-Karlsson, P. Petersson, P. Spagel and S. Nilsson, Anal. Chem., 2002, 74, 4595–4601 CrossRef CAS.
- C. Nilsson, P. Viberg, P. Spegel, M. Jornten-Karlsson, P. Petersson and S. Nilsson, Anal. Chem., 2006, 78, 6088–6095 CrossRef CAS.
- B. Neiman, E. Grushka, J. Gun and O. Lev, Anal. Chem., 2002, 74, 3484–3491 CrossRef CAS.
- M. R. Ivanov, H. R. Bednar and A. J. Haes, ACS Nano, 2009, 3, 386–394 CrossRef CAS.
- L. Yang, E. Guihen, J. D. Holmes, M. Loughran, G. P. O'Sullivan and J. D. Glennon, Anal. Chem., 2005, 77, 1840–1846 CrossRef CAS.
- Z. Zhang, B. Yan, K. Liu, Y. Liao and H. Liu, Electrophoresis, 2009, 30, 379–387 CrossRef CAS.
- S. Bachmann, Electrophoresis, 2010, 31, 618–629 CrossRef CAS.
- U. Schnabel, C. Fischer and E. Kenndler, J. Microcolumn Sep., 1997, 9, 529–534 CrossRef CAS.
- U. Pyell, Electrophoresis, 2010, 31, 814–831 CrossRef CAS.
- M. Hanauer, P. Sebastien, I. Zins and A. Lotz, Nano Lett., 2007, 7, 2881–2885 CrossRef CAS.
- W. Bucking and T. Nann, IEE Proc.: Nanobiotechnol., 2006, 153, 47–53 CrossRef CAS.
- S. K. Doorn, M. S. Strano, M. J. O'Connell, E. H. Haroz, K. L. Rialon, R. H. Hauge and R. E. Smalley, J. Phys. Chem. B, 2003, 107, 6063–6069 CrossRef CAS.
- C. K. Lo, M. C. Paau, D. Xiao and M. M. F. Choi, Anal. Chem., 2008, 80, 2439–2446 CrossRef CAS.
- S. Radko and A. Chrambach, Electrophoresis, 2002, 23, 1957–1972 CrossRef CAS.
- F. K. Liu, F. H. Ko, P. W. Huang, C. H. Wu and T. C. Chu, J. Chromatogr., A, 2005, 1062, 139–145 CrossRef CAS.
- H. K. Jones and N. E. Ballou, Anal. Chem., 1990, 62, 2484–2490 CrossRef CAS.
- F. Liu, Anal. Chim. Acta, 2005, 528, 249–254 CrossRef CAS.
- F. d'Orlye, A. Varenne, T. Georgelin, J. Siaugue, B. Teste, S. Descroix and P. Gareil, Electrophoresis, 2009, 30, 2572–2582 CrossRef CAS.
- S. Zhao, T. Niu, Y. Song and Y. Liu, Electrophoresis, 2009, 30, 1059–1065 CrossRef CAS.
- C. Nilsson, K. Becker, I. Harwigsson, L. Bullow, S. Birnbaum and S. Nilsson, Anal. Chem., 2009, 81, 315–321 CrossRef CAS.
- C. Nilsson, I. Harwigsson, K. Becker, J. P. Kutter, S. Birnbaum and S. Nilsson, Electrophoresis, 2010, 31, 459–464 CrossRef CAS.
- M. Lucarini, P. Franchi, G. F. Pedulli, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2004, 126, 9326–9329 CrossRef CAS.
- M. Lucarini, P. Franchi, G. F. Pedulli, C. Gentilini, S. Polizzi, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2005, 127, 16384–16385 CrossRef CAS.
- E. A. S. Doherty, R. J. Meagher, M. N. Albarghouthi and A. E. Barron, Electrophoresis, 2003, 24, 34–54 CrossRef CAS.
- J. Horvath and V. Dolník, Electrophoresis, 2001, 22, 644–655 CrossRef CAS.
- J. E. Melanson, N. E. Baryla and C. A. Lucy, Anal. Chem., 2000, 72, 4110–4114 CrossRef CAS.
- C. Wang and C. A. Lucy, Anal. Chem., 2005, 77, 2015–2021 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |