DOI:
10.1039/C0AN00439A
(Paper)
Analyst, 2011,
136, 148-154
Luminescent ruthenium probe for the determination of acetyl phosphate in complex biological matrices†
Received
24th June 2010
, Accepted 20th September 2010
First published on 18th October 2010
Abstract
The first probe for the fluorogenic determination of acetyl phosphate (AcP), (bpy)2Ru(1,10-phenanthroline-5,6-dione dioxime) (RuPDO), was prepared and its reaction with AcP was studied in detail. The emission of the weakly luminescent RuPDO is red shifted and strongly enhanced upon reaction with AcP in the presence of metal cations like Zn2+ or Cu2+. The reaction occurs within 60 min incubation time under highly biocompatible conditions (aqueous buffer of pH 7, 37 °C). A linear dynamic range from 10 to 200 µmol L−1 is observed with an LOD of AcP of 3.4 µmol L−1 (for RuPDO-Zn). Other bio-phosphates studied show only weak interference. Furthermore, the applicability of the probe in complex biological matrices was evaluated.
Introduction
Acetyl phosphate (AcP) is a high energy metabolic compound derived from acetic acid and orthophosphoric acid. It was first found by Lipmann in the early forties of the last century1 leading to the discovery of coenzyme A.2Acetyl phosphate was also proposed as a primeval form of metabolic energy currency due to its simple chemical structure and high phosphorylation potential (ΔG0 (AcP): −43 kJ mol−1 and ΔG0 (ATP): −31 kJ mol−1).3–5 Therefore, AcP is primarily found in prokaryotes to serve as a regulatory signal transducer, i.e. in the ATP-dependent proteolysis,6activation of the phosphate regulation,7 in motility and chemotaxis expression,8 or in the initiation of membrane phospholipid synthesis9 of countless microorganisms. The steady state concentration of AcP in wild type E. coli reaches at least 3 mmol L−1(ref. 10) and Wolfe et al.11 proved that acetyl phosphate acts as a global sign in E. coli. AcP is also a non-nucleotide substrate for the Ca-ATPase12 and an important source for regeneration of ATP.7 Recently, the presence of an acetate kinase-phosphotransacylase-pathway (involving acetyl phosphate) was also validated for some eukaryotic microbe species.13 Up to date, there is no clear evidence that AcP is a stable metabolic intermediate in higher living organisms. On the one hand, enzymes permitting the production of acetyl phosphate from citric acid for example are found in animal tissues.14 On the other hand, also highly active acetylphosphatase is present in these tissues.15 Furthermore, acetate—the prokaryotic metabolic product of AcP breakdown—is generated endogenously in mammalian cells from acetyl-CoA hydrolase without using AcP.16 There might also be a chance of retrieving acetyl phosphate in those cells considering the Warburg hypothesis17 that tumor cells generate their energy mainly by non-oxidative breakdown of glucose followed by lactic acid fermentation. In 2009, this hypothesis could be confirmed with VA13 cells by Engler et al.18 Further assuming that non-malign cells do not produce and preserve AcP in high concentrations, it might be a powerful tumor marker. AcP attracted some attention in the commercial synthesis of ATPviaacetate kinase. This system also provides a cheap and stable source for regeneration of ATP in bacterial batch reactors.19,20
Up to date, the most common methods for the determination of acetyl phosphate are the colorimetric hydroxylaminolysis and some acetate kinase (AcK) coupled enzymatic methods. The hydroxamate assay was originally developed by Lipmann and Tuttle21 and frequently optimized, among others, by Rose22 and Pechère and Capony.23 Briefly, AcP is reacted with hydroxylamine and the hydroxamic acid formed reacts with ferric ions to form an orange colored complex. This assay is simple and fast but requires strongly acidic pH and high concentrations of hydroxylamine (704 mM). This is not suitable for direct analysis of biological samples. Its limit of detection (LOD) is about 40 µmol L−1. Recently, this assay was coupled to an enzymatic reaction overcoming some of these limitations.24AcP is hydrolyzed by lowly concentrated hydroxylamine. The inorganic phosphate produced is then determined with a commercially available kit25 using a purine nucleoside phosphorylase to yield a product absorbing at 330 nm at µmolar concentrations of AcP. The main drawback of this assay is that the presence of high concentrations of inorganic phosphate (Pi) in samples prevents direct AcP determination. Further on, Prüß and Wolfe26 presented an AcK-based assay wherein the ATP formed is determined by the Luciferase reaction.27 This assay was further improved by Ito et al.28 and Zhao et al.29 Another sensitive, yet time consuming method utilizing AcK involves the conversion of acetyl phosphate with radioactive [3H]ADP to [3H]ATP, separation by thin layer chromatography (TLC) and scintillation signal readout.30 As low as 20 nmol L−1 of AcP were determined. Furthermore, AcP was assayed by supplementing cells with [32Pi] and 2D-TLC of the cell extract.31 Most of these methods share the characteristics that either they are conducted under non-biocompatible conditions or that they are complicated and time consuming.
Our aim was to develop a luminescent method for the direct determination of AcP under biocompatible conditions. We intended to transfer the tunable (e.g. by the addition of Zn, Cu, Ni, and Co) and selective reaction of hydroxylamines or hydroxamic acids32,33 to a luminescent reporter molecule. Ruthenium complexes are known to have interesting spectral properties, e.g. large Stokes' shift, high photostability and good quantum yields. Hence, we found (bpy)2Ru(1,10-phenanthroline-5,6-dione dioxime) (RuPDO) to be an interesting candidate for further investigations. We show that upon reaction with AcP in the presence of Zn2+ or Cu2+ a strong increase in luminescence intensity is accompanied by a bathochromic shift. This selective reaction occurs under highly biocompatible conditions and enables determination of AcP down to the low µmol L−1 range. Although RuPDO does not emit from an excited singlet state, we refer to it as a fluorogenic probe as this is a much more common term than a luminogenic probe. Furthermore, we show the high potential of RuPDO for determination of acetyl phosphate in complex biological matrices.
Experimental
Instrumentation
Absorption spectra were recorded on a Cary 50 Bio UV-Vis Spectrophotometer (Varian, Australia, http://www.varianinc.com/). Luminescence spectra were recorded on an Aminco-Bowman AB 2 luminescence spectrometer (http://www.thermo.com/) equipped with a 150 W continuous wave xenon lamp as excitation light source. All spectra are uncorrected. Lifetime measurements were done on an ISS K2 multifrequency cross-correlation phase modulation fluorimeter (http://www.iss.com/) using an argon ion laser for excitation. Microtiter plate experiments were performed on a Tecan Genios Plus Reader (http://www.tecan.de/) at λexc = 485 nm and λem = 635 nm in black flat bottom 96 well plates from Greiner Bio-One (http://www.gbo.com/). pH was measured with a pH meter CG 842 from Schott (http://www.schott.com/) at room temperature. The ESI mass spectra were taken on a ThermoQuest TSQ 7000 (http://www.thermo.com/) mass spectrometer. IR spectra of solids were recorded with a diamond ATR-crystal on a Varian 670-IR spectrometer (http://www.varianinc.com/).
Materials
cis-Dichlorobis(2,2′-bipyridine)ruthenium(II) dihydrate (Ru(bpy)2Cl2) was purchased from ABCR (http://www.abcr.de/). All other chemicals and solvents were purchased from Sigma Aldrich (http://www.sigmaaldrich.com/) or Acros Organics (http://www.acros.be/). Stock solutions of acetyl phosphate (10.0 or 1.0 mmol L−1) were prepared in HEPES buffer of pH 7.4 (40 mmol L−1) shortly before measurements. Stock solutions of RuPDO (1.0 mmol L−1) were prepared by pre-dissolving of 0.94 mg of the reagent in 10 µL of DMSO followed by dilution with HEPES buffer to 1.0 mL. The RuPDO stock solution was stored at 4 °C. Stock solutions of ZnCl2 were prepared by dissolving 34.1 mg of the reagent in 50 mL of HEPES buffer.
Synthesis of RuPDO
RuPDO was synthesized with slight modifications according to the literature procedure34,35 in good yields. Ru(bpy)2Cl2 (74 mg) was dissolved in dry ethanol (10 mL), then 1,10-phenanthroline-5,6-dione (phendione) (29 mg) was added and the violet solution heated to reflux for 5 h under N2. After cooling to room temperature, the solvent was removed viarotary evaporation. Ru(bpy)2(phendione) was redissolved in ethanol (15 mL), CaCO3 (dried, 34 mg) added and the mixture was heated to 81 °C. A solution of NH2OH ·HCl (53 mg) in 5 mL of ethanol was added dropwise over a period of 1 h, while the orange solution turned to a reddish color. After reflux for 5 h and cooling to room temperature the solvent was evaporated. The remaining orange to red colored solid was dissolved in a small amount of H2O and the complex was precipitated by adding excess of NH4PF6 (Scheme 1). The red precipitate was washed with few mL of water and diethyl ether. The crude product (60% yield) was recrystallized from acetone (50% yield). Found: C 38.85, H 3.21, N 11.51. C32H24F12N8O2P2Ru·3H2O requires C 38.53, H 3.03, N 11.23%; m/z (ESI) 326.8 (M2+, C32H24N8O2Ru requires 327.1).
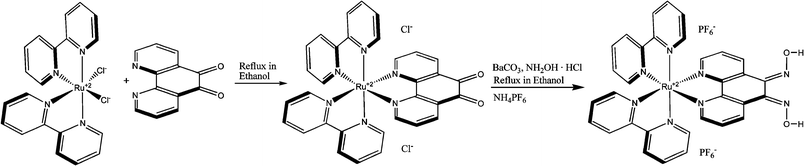 |
| | Scheme 1 Synthesis of RuPDO. | |
Protocol for determination of AcP
50 µL of the RuPDO stock solution (final concentration 50 µmol L−1 in 40 mmol L−1HEPES buffer of pH 7.4) and 100 µL of ZnCl2 (final concentration 0.5 mmol L−1) were added to HEPES buffer of pH 7.4 (40 mmol L−1) containing at least 0.05 mmol L−1 of AcP and made up to 1 mL (for tests in cuvettes). One-tenth of all volumes were used for microtiter plate based experiments (final volume 0.1 mL). The same amount of RuPDO and ZnCl2 was added to the control containing no AcP. The reaction vials were then kept at 37 °C for 60 min before measuring luminescence.
Results and discussion
Spectral properties and response to AcP
A solution of RuPDO in aqueous buffer shows only weak luminescence (λexc/em at 469/610 nm). For determination of acetyl phosphate, the vic-oxime groups of the phenanthroline ligand are activated in situ by the addition of cations like Zn2+, Cu2+ or Ni2+. The addition of these activators does not alter the emission spectrum of RuPDO. The luminescence spectra of RuPDO with (RuPDO-Zn) and without activator cation (RuPDO) and in the presence of AcP (AcP-RuPDO-Zn) are shown in Fig. 1.
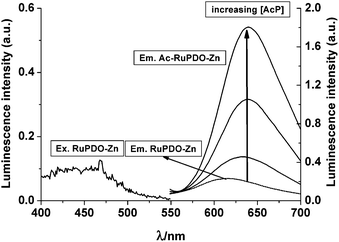 |
| | Fig. 1
Excitation spectrum of RuPDO-Zn and emission spectra of RuPDO-Zn and the reaction product with AcP (AcP-RuPDO-Zn) after addition of increasing concentrations of AcP: 50 µmol L−1RuPDO, 0.5 mmol L−1Zn2+, 20 µmol L−1, 0.1 mmol L−1, 0.5 mmol L−1 of AcP, respectively, 40 mmol L−1HEPES buffer of pH 7.4, 60 min incubation at 37 °C. The emission spectrum of RuPDO closely matches the one of RuPDO-Zn and is almost invisible. | |
A red shift of the emission of RuPDO of 30 nm (from 610 nm to 640 nm) is observed in the presence of AcP. This is also accompanied by a strong enhancement of fluorescence intensity, increase of quantum yield and change of decay time (see Table 1). The wavelength of the excitation maximum remains unchanged. These spectroscopic changes solely occur in the presence of transition metal cations and active acetic acid esters like AcP or 4-nitrophenyl acetate (4 NA). This effect is not observed in the presence of Ca2+ or Mg2+ (data not shown). Hence, a proposed reaction mechanism is shown in Scheme 2 and evidence for the mechanism is given in a later section.
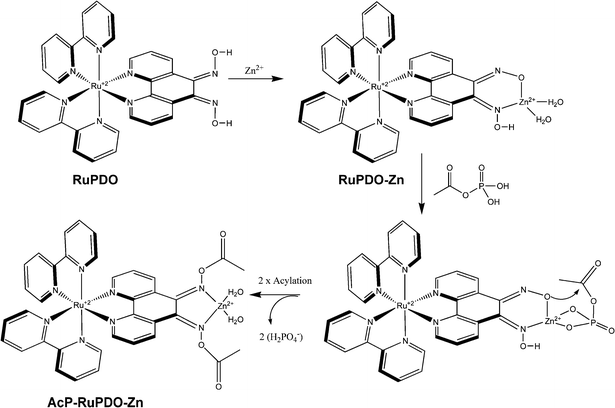 |
| | Scheme 2 Reaction mechanism of RuPDO-Zn with AcP. | |
Table 1 Spectral properties of RuPDO and the reaction product (AcP-RuPDO-Zn)
| |
RuPDO
|
AcP-RuPDO-Zn
|
|
In air saturated ethanol; reference: Rhodamine 6G (Φ = 0.95).
In air saturated HEPES buffer (40 mmol L−1, pH 7.4).
|
|
λ
exc/nm |
469 |
469 |
|
λ
em/nm |
610 |
640 |
|
ε
430, ε469/L mol−1 cm−1 |
7200, 5800 |
6300, 4400 |
|
Φ
a
|
0.64% |
4.5% |
|
τ
b
|
τ
1: 351 ns (81.6%) |
τ
1: 280 ns (19.6%) |
|
τ
2: 81 ns (16.3%) (χ2 = 2.5) |
τ
2: 149 ns (80.4%) (χ2 = 2.1) |
A further benefit of RuPDO is the large Stokes' shift of 141 nm (RuPDO-Zn) and 171 nm (AcP-RuPDO-Zn), respectively. This warrants excellent blocking of excitation light in filter-based fluorescence instrumentation. The fairly strong red fluorescence of AcP-RuPDO-Zn also permits measurements in biological samples with almost zero background. Furthermore, the reaction product can easily be distinguished from the probe itself as the former shows much stronger and red shifted (by 30 nm) emission.
The longwave emission of AcP-RuPDO-Zn suggests further investigations for detection of AcPin vivo, where emission at >600 nm is a prerequisite to avoid autofluorescence from tissue and warrants suitable penetration depth (due to reduced absorption of tissue).
Choice of activating cation
Several cations were screened for their oxime activation properties (Table 2). The reaction of RuPDO with AcP was carried out in HEPES buffer, pH 7.4 (40 mmol L−1), at 37 °C in the presence of 1 mmol L−1 of AcP, 0.5 mmol L−1 of activating cation and 50 µmol L−1 of RuPDO. The luminescence intensity at 640 nm was referenced with respect to the signal found for Zn2+-activation at 640 nm after 60 min incubation time. Finally, only Zn2+ and Cu2+ were selected for further investigations due to their higher biocompatibility compared to the other cations tested and the strong response to AcP.
Table 2 Effect of activating cations
| Activator cation |
I
M
x+/IZn2+a |
Activator cation |
I
M
x+/IZn2+a |
|
After 60 min incubation time, each.
|
|
Zn2+ |
100% |
Ce4+ |
17% |
|
Cu+ |
52% |
Ce3+ |
66% |
|
Cu2+ |
87% |
La3+ |
39% |
|
Cd2+ |
51% |
Ni2+ |
86% |
|
Pd2+ |
13% |
Yb3+ |
54% |
|
Ru3+ |
13% |
Y3+ |
58% |
|
Cr2+ |
13% |
Tb3+ |
53% |
|
Ag+ |
18% |
Eu3+ |
46% |
|
Co2+ |
14% |
|
|
Effect of pH and reaction time
The effect of pH on the reaction of Zn2+- and Cu2+-activated RuPDO with AcP was investigated via luminescence measurement at 640 nm. The pH dependence of the reaction of RuPDO-Zn and RuPDO-Cu with AcP is shown in Fig. 2. It is in good correlation to previous data of Cu2+ and Zn2+ activated oximes.36,37 It is possible to determine AcP at physiological pH with RuPDO-Zn as well as with RuPDO-Cu. Therefore, pH 7.4 was chosen as the optimal pH for all further investigations.
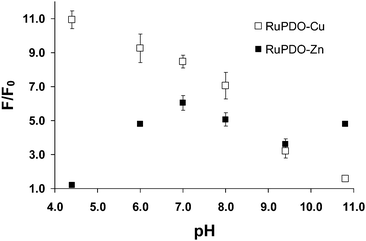 |
| | Fig. 2 Effect of pH on the reaction of RuPDO-Zn/Cu with AcP. 10 mmol L−1 piperacine–glycylglycine buffer, 50 µmol L−1RuPDO, 0.5 mmol L−1ZnCl2 or CuCl2, respectively, 1 mmol L−1AcP, 60 min incubation at 37 °C (n = 4). | |
The reaction rate of 50 µmol L−1RuPDO in the presence of 0.5 mmol L−1Zn2+ or Cu2+ with 0.1, 0.2 and 1 mmol L−1 of AcP, respectively, was also examined at 37 °C viaF/F0 at 640 nm (Fig. 3). After 60 min incubation, over 90% of the maximum signal is achieved at all concentrations of AcP. Therefore, a 1 h incubation time was chosen for all further investigations.
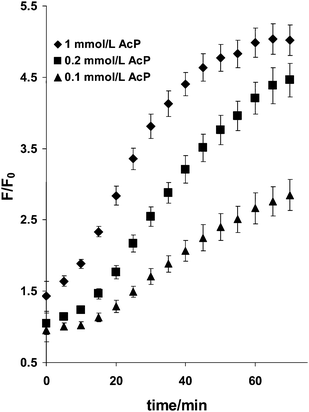 |
| | Fig. 3 Effect of reaction time on the reaction of RuPDO-Zn with AcP. 40 mmol L−1HEPES buffer of pH 7.4, 50 µmol L−1RuPDO, 0.5 mmol L−1Zn2+, 0.1, 0.2, and 1 mmol L−1 of AcP, respectively, 60 min reaction at 37 °C (n = 4). | |
Mechanism of fluorogenic reaction
We were further interested in the mechanism of the reaction between RuPDO-Zn and AcP. The proposed reaction mechanism (Scheme 2) was inspired by the works of Lipmann and Tuttle,21 Weijnen et al.38 and Mancin et al.39 The two main possibilities clearly are either an acetylation of the oxime groups or a phosphorylation. Therefore, we added 4-nitrophenyl acetate (4 NA) to a buffered solution of RuPDO-Zn and in another experiment bis(nitrophenyl) phosphate (BNPP). In case of 1 mmol L−1 4 NA, the same increase of luminescence intensity peaking at 640 nm was observed as with AcP solution of equal concentration. Additionally, an increase of the absorbance at 400 nm was found due to absorption of the 4-nitrophenolate anion formed during the reaction. Neither the emission spectrum nor the absorption spectrum was affected during reaction of RuPDO-Zn with BNPP or other biologically relevant phosphates like inorganic phosphate (Pi), pyrophosphate (PP), ATP and ADP. This is a clear evidence for the oxime acetylation thesis. Furthermore, the IR spectra of RuPDO prior and after reaction with AcP (AcP-RuPDO-Zn) differ mainly due to the appearance of the new aliphatic C–H stretching vibrations at 2923 and 2853 cm−1, respectively, of the O–Ac group (AcP-RuPDO-Zn).
Complex stoichiometry and calibration plots
The stoichiometry of the reaction was determined via Job's method of continuous variation. From Fig. 4, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the reaction of RuPDO-Zn and AcP under the given reaction conditions can be deduced. However, one can assume that in the presence of excess acetyl phosphate both oxime groups of RuPDO will be involved in the reaction. This behavior was observed in the calibration plots in Fig. 5. The fact that linearity of the calibration plot exceeds equimolar concentrations between AcP and RuPDO suggests the participation of the second oxime reaction site. A linear dynamic range from 10 to 200 µmol L−1 of AcP and 80 to 400 µmol L−1 of AcP at a probe concentration of 50 µmol L−1 is observed for RuPDO-Zn and RuPDO-Cu, respectively. The regression equation for RuPDO-Zn is F/F0 = 16.59 L mmol−1 [AcP] + 1.29 (r = 0.990) and F/F0 = 4.28 L mmol−1 [AcP] + 0.74 (r = 0.990) for RuPDO-Cu. The limit of detection of AcPviaRuPDO-Zn is 3.4 µmol L−1. Hence, AcP can be determined at least at 10 times lower concentrations compared to the method of Lipmann and Tuttle.21
:1 stoichiometry of the reaction of RuPDO-Zn and AcP under the given reaction conditions can be deduced. However, one can assume that in the presence of excess acetyl phosphate both oxime groups of RuPDO will be involved in the reaction. This behavior was observed in the calibration plots in Fig. 5. The fact that linearity of the calibration plot exceeds equimolar concentrations between AcP and RuPDO suggests the participation of the second oxime reaction site. A linear dynamic range from 10 to 200 µmol L−1 of AcP and 80 to 400 µmol L−1 of AcP at a probe concentration of 50 µmol L−1 is observed for RuPDO-Zn and RuPDO-Cu, respectively. The regression equation for RuPDO-Zn is F/F0 = 16.59 L mmol−1 [AcP] + 1.29 (r = 0.990) and F/F0 = 4.28 L mmol−1 [AcP] + 0.74 (r = 0.990) for RuPDO-Cu. The limit of detection of AcPviaRuPDO-Zn is 3.4 µmol L−1. Hence, AcP can be determined at least at 10 times lower concentrations compared to the method of Lipmann and Tuttle.21
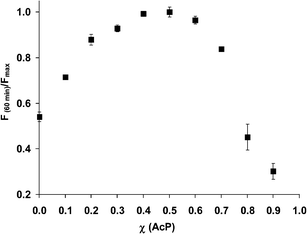 |
| | Fig. 4 Normalized fluorescence at 640 nm vs. mole fraction AcP. Data collected after 60 min of incubation at 37 °C in 40 mmol L−1HEPES of pH 7.4 (n = 4). | |
![Plot of F/F0vs. [AcP] collected after 60 min of incubation at 37 °C in 40 mmol L−1HEPES of pH 7.4 (n = 4).](/image/article/2011/AN/c0an00439a/c0an00439a-f5.gif) |
| | Fig. 5 Plot of F/F0vs. [AcP] collected after 60 min of incubation at 37 °C in 40 mmol L−1HEPES of pH 7.4 (n = 4). | |
Interference of other relevant phosphates
The reaction of RuPDO-Zn with AcP in the presence of equimolar concentrations of inorganic phosphate (Pi), pyrophosphate (PP) and ATP was monitored using luminescence (Fig. 6). These phosphates slightly quench the luminescence of RuPDO-Zn, probably due to coordination to Zn2+. Within a tolerance limit of ±10% the following concentrations of interferents are acceptable: Pi = 1 mmol L−1, PP = 0.22 mmol L−1 and ATP = 0.67 mmol L−1. Hence, even in the presence of equimolar concentrations of AcP and phosphate (1 mmol L−1 as found in DMEM medium, see next section) a pronounced increase of luminescence is detectable. As a result, the slope of a calibration plot for AcP in the presence of equimolar phosphate is slightly reduced. We assume that binding of these phosphates to RuPDO-Zn blocks the “binding-site” for AcP, resulting in a lower acetylation-rate and therefore a lower luminescence. Addition of phosphate to a reacted solution (90 min) of AcP-RuPDO-Zn does not change the signal (data not shown), substantiating the binding-site-blocking thesis. The binding of phosphates to the probe can adequately be reduced by adding a higher amount of Zn2+ to the sample, resulting in higher luminescence intensity due to complexation of phosphate by free Zn2+ in solution (Fig. S1†). It is obvious from Fig. S1† that higher concentrations of Zn2+ are able to overcome the quenching-effect of Pi.
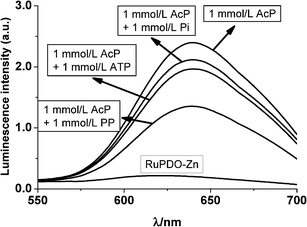 |
| | Fig. 6 Effect of bio-phosphates on the luminescence of the reaction of RuPDO-Zn with AcP. 40 mmol L−1HEPES buffer of pH 7.4, 50 µmol L−1RuPDO, 0.5 mmol L−1Zn2+, 1 mmol L−1 of AcP and inorganic phosphate (Pi), pyrophosphate (PP), ATP, each, 60 min reaction at 37 °C (n = 4). | |
Tests of RuPDO-Zn in cell medium containing NRK cells
The reaction of RuPDO-Zn with AcP was examined in Dulbecco's Modified Eagle Medium (DMEM) and also in DMEM medium containing normal rat kidney (NRK) cells in order to test the future applicability of the probe for measurements in biological matrices. DMEM medium used in the experiments contains, among others, high millimolar concentrations of inorganic compounds like CaCl2, NaH2PO4 and KCl, various amounts of different amino acids and 10% (w/w) fetal calf serum. NRK cells were grown in DMEM medium for 2 days. During this period the medium is further enriched with products of cell metabolism. Hence, the cell medium used is a complex matrix and served as reaction medium without further workup. The medium together with 50 µmol L−1 of RuPDO, 0.5 mmol L−1ZnCl2 and various amounts of AcP was incubated at 37 °C and the progress of the reaction was monitored for 90 min by recording the increase of the luminescence intensity at 640 nm. An almost 2.5 fold signal increase in both DMEM medium containing NRK cells (Fig. 7) and medium without cells (data not shown) was observed on addition of 2 mmol L−1 of AcP. As a matter of fact, the signal increase is not as high as observed in plain aqueous buffered solution due to the complex matrix. The slope is reduced by a factor of 20 compared to aqueous buffer. However, it is obvious from the calibration plot that determination of AcP from 0.5 to 2 mmol L−1 is possible with reasonable precision in complex biological matrices using the luminescence of RuPDO-Zn. Importantly, the reaction time in cell medium is much shorter compared to the reaction in aqueous buffer. The maximum luminescence intensity at 640 nm is obtained after 30 min of incubation. This strongly supports the proposed reaction mechanism of RuPDO-Zn. A high amount of RuPDO-Zn is already blocked in the presence of high concentrations of phosphate. Therefore, only few RuPDO-Zn molecules are able to react with AcP, resulting in an decreased luminescence intensity at 640 nm and shorter reaction time compared to a sample in plain buffer.
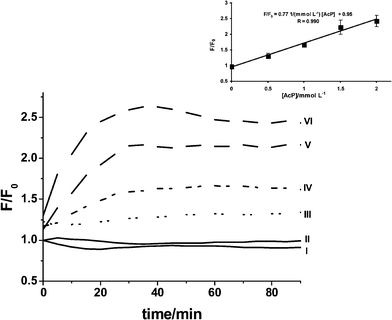 |
| | Fig. 7 Luminescence of RuPDO-Zn in suspensions of NRK cells in DMEM cell medium after spiking with AcP. Solid lines (I and II) represent unspiked samples without cells (I) and in the presence of cells (II). Dashed lines (III to VI) represent spiked samples of cell media containing NRK cells. AcP concentration is increasing from 0.5 mmol L−1 (III) to 2 mmol L−1 (VI) (n = 4). | |
Additional tests of RuPDO-Zn were conducted in E. colilysogeny broth (LB) media without bacteria, in LB media containing E. coli, and in LB media containing sonicated E. coli (see ESI†). The kinetic of the reaction of RuPDO-Zn with AcP in E. coli LB media without bacteria is shown in Fig. S2†. The reaction of RuPDO-Zn with AcP is slower than in DMEM medium (Fig. 7). While the increase of luminescence intensity is slower, the slope of the calibration plot is steeper (Fig. S3†) than in DMEM medium (inset of Fig. 7). The kinetic of the reaction of RuPDO-Zn with AcP in LB media containing E. coli is shown in Fig. S4† and remains almost unchanged with respect to LB medium without E. coli (Fig. S2†). The increase of luminescence intensity is lower, resulting in a lower slope of the calibration plot (Fig. S5†) than in LB medium without E. coli (Fig. S3†). This behavior could be expected because cells usually show strong scatter in luminescence measurements. The kinetic of the reaction of RuPDO-Zn with AcP in LB media containing sonicated E. coli is shown in Fig. S6† and remains almost unchanged with respect to LB medium containing E. coli (Fig. S4†). Here, the increase of luminescence intensity (Fig. S7†) is almost constant with respect to the unlysed cells (Fig. S5†). These data clearly show the applicability of the probe in various complex biological matrices.
Conclusions
The first fluorogenic probe RuPDO is introduced for the rapid and direct determination of acetyl phosphate in aqueous solutions at neutral pH. Unlike in common methods for determination of AcP, the presence of millimolar inorganic phosphate only weakly reduces sensitivity and no radioactive waste is produced. Successful determination of AcP in complex biological matrices and the longwave emission of AcP-RuPDO-Zn suggest further investigations for detection of AcPin vivo. A combination of RuPDO with suitable enzymes involved in the formation or conversion of AcP suggests luminescent probing of other biological important analytes like glucose, α-ketoglutarate, and acetate. These experiments are now under way.
Acknowledgements
MSS gratefully acknowledges financial support from the BMBF (German Ministry for Education and Research) within the Biophotonik II programme for the joint project “TumorVision”. We thank Barbara Goricnik from the Bioanalytics and Biosensors group (Prof. Dr. J. Wegener) of our institute for conducting the cell growth experiments.
References
- F. Lipmann and C. L. Tuttle, J. Biol. Chem., 1944, 153, 571–582 CAS.
- N. Kresge, R. D. Simoni and R. L. Hill, J. Biol. Chem., 2005, 280, 164–166 CAS.
- J. G. Ferry and C. H. House, Mol. Biol. Evol., 2006, 23, 1286–1292 CrossRef CAS.
- C. de Duve, Origins Life Evol. Biosphere, 2003, 33, 559–574 CrossRef CAS.
- W. Martin and M. J. Russell, Philos. Trans. R. Soc. London, Ser. B, 2007, 362, 1887–1926 CrossRef CAS.
- I. Mizrahi, D. Biran and E. Z. Ron, Mol. Microbiol., 2006, 62, 201–211 CrossRef CAS.
- S. K. Kim, M. R. Wilmes-Riesenberg and B. L. Wanner, Mol. Microbiol., 1996, 22, 135–147 CAS.
- I. Gueriri, S. Bay, S. Dubrac, C. Cyncynatus and T. Msadek, Mol. Microbiol., 2008, 70, 1342–1357 CrossRef CAS.
- Y. J. Lu, Y. M. Zhang, K. D. Grimes, J. Qi, R. E. Lee and C. O. Rock, Mol. Cell, 2006, 23, 765–772 CrossRef CAS.
- A. H. Klein, A. Shulla, S. A. Reimann, D. H. Keating and A. J. Wolfe, J. Bacteriol., 2007, 189, 5574–5581 CrossRef CAS.
- A. J. Wolfe, Curr. Opin. Microbiol., 2010, 13, 204–209 CrossRef CAS.
- F. Soler, M. I. Fortea, A. Lax and F. Fernández-Belda, J. Biol. Chem., 2002, 277, 38127–38132 CrossRef CAS.
- C. Ingram-Smith, S. R. Martina and K. S. Smitha, Trends Microbiol., 2006, 14, 249–253 CrossRef CAS.
- M. F. Guly, T. N. Pechenova and L. I. Matusevich, Nature, 1966, 212, 36–37 CrossRef CAS.
- B. Shapiro and E. Wertheimer, Nature, 1945, 156, 690 CrossRef CAS.
- B. Crabtree, M. J. Souter and S. E. Anderson, Biochem. J., 1989, 257, 673–678 CAS.
- O. Warburg, F. Wind and E. Negelein, J. Mol. Med., 1926, 5, 829–832 CAS.
- I. Engler, C. Atzmüller, V. Donic and F. Steinhäusler, J. Exp. Ther. Oncol., 2009, 8, 157–165 CAS.
- R. L. Baughn, O. Adalsteinsson and G. M. Whitesides, J. Am. Chem. Soc., 1987, 100, 304–306.
- L. A. Ryabova, L. M. Vinokurov, E. A. Shekhovtsova, Y. B. Alakhov and A. S. Spirin, Anal. Biochem., 1995, 226, 184–186 CrossRef CAS.
- F. Lipmann and L. C. Tuttle, J. Biol. Chem., 1945, 159, 21–28 CAS.
- I. A. Rose, Methods Enzymol., 1955, 1, 591–595 Search PubMed.
- J. F. Pechère and J. P. Capony, Anal. Biochem., 1968, 22, 536–539 CrossRef CAS.
- S. Mukhopadhyay, M. S. Hasson and D. A. Sanders, Bioorg. Chem., 2008, 36, 65–69 CAS.
- M. R. Webb, Proc. Natl. Acad. Sci. U. S. A., 1992, 1, 4884–4887 CrossRef.
- B. M. Prüß and A. J. Wolfe, Mol. Microbiol., 1994, 12, 973–984 CrossRef.
- A. Lundin and A. Thore, Anal. Biochem., 1975, 66, 47–63 CrossRef CAS.
- K. Ito, K. Nakagawa, S. Murakami, H. Arakawa and M. Maeda, Anal. Sci., 2003, 19, 105–109 CAS.
- Y. Zhao, C. A. Tomas, F. B. Rudolph, E. T. Papoutsakis and G. N. Bennett, Appl. Environ. Microbiol., 2005, 71, 530–537 CrossRef CAS.
- A. G. Hunt and J. S. Hong, Anal. Biochem., 1980, 108, 290–294 CAS.
- D. H. Keating, A. Shulla, A. H. Klein and A. J. Wolfe, Biol. Proced. Online, 2008, 10, 36–46 Search PubMed.
- C. H. Oestreich and M. M. Jones, Biochemistry, 1966, 5, 2926–2931 CrossRef CAS.
- P. J. Briggs, D. P. N. Satchell and G. F. White, J. Chem. Soc. B, 1970, 1008–1012 RSC.
- S. Bodige and F. M. MacDonnell, Tetrahedron Lett., 1997, 38, 8159–8160 CrossRef CAS.
- M. Li, Z. Xiao, Z. Huan and Z. Lu, Appl. Surf. Sci., 1998, 125, 217–220 CrossRef CAS.
- F. Mancin, P. Tecilla and U. Tonellato, Langmuir, 2000, 16, 227–233 CrossRef CAS.
- S. Liu and A. D. Hamilton, Tetrahedron Lett., 1997, 38, 1107–1110 CrossRef CAS.
- J. G. J. Weijnen, A. Koudijs, G. A. Schellekens and J. F. J. Engbersen, J. Chem. Soc., Perkin Trans. 2, 1992, 829–834 RSC.
- F. Mancin, P. Tecilla and U. Tonellato, Langmuir, 2000, 16, 227–233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Effect of Zn-concentration on luminescence intensity; testing of RuPDO in different complex biological matrices. See DOI: 10.1039/c0an00439a |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.