Time of flight mass spectrometry for quantitative data analysis in fast transient studies using a Temporal Analysis of Products (TAP) reactor†
Alexandre
Goguet
*a,
Christopher
Hardacre
a,
Noleen
Maguire
a,
Kevin
Morgan
a,
Sergiy O.
Shekhtman
*a and
Steve P.
Thompson
b
aCenTACat, School of Chemistry and Chemical Engineering, Queen's University, Stranmillis Road, Belfast, BT9 5AG, Northern Ireland, UK. E-mail: a.goguet@qub.ac.uk; s.shekthman@qub.ac.uk
bScientific Analysis Instruments Ltd, Hadfield House, Hadfield Street, Old Trafford, Manchester, M16 4FE, UK
First published on 4th October 2010
Abstract
A Time of flight (ToF) mass spectrometer suitable in terms of sensitivity, detector response and time resolution, for application in fast transient Temporal Analysis of Products (TAP) kinetic catalyst characterization is reported. Technical difficulties associated with such application as well as the solutions implemented in terms of adaptations of the ToF apparatus are discussed. The performance of the ToF was validated and the full linearity of the specific detector over the full dynamic range was explored in order to ensure its applicability for the TAP application. The reported TAP-ToF setup is the first system that achieves the high level of sensitivity allowing monitoring of the full 0–200 AMU range simultaneously with sub-millisecond time resolution. In this new setup, the high sensitivity allows the use of low intensity pulses ensuring that transport through the reactor occurs in the Knudsen diffusion regime and that the data can, therefore, be fully analysed using the reported theoretical TAP models and data processing.
Introduction
The non-steady-state kinetic characterization of catalysts, especially industrial multi-component catalysts, is an extremely important focus of heterogeneous catalytic research. Without such characterization the non-steady-state performance of different catalysts (activity and selectivity) cannot be compared, a dynamic kinetic model of the catalytic transformations cannot be developed, and finally, efficient catalytic reactors that work under non-steady-state conditions cannot be designed.In a typical kinetic characterization experiment, the heterogeneous catalytic process involves both complex chemical reactions and transport. Transport is also a complex process, which generally includes both convection and diffusion. The essential property of an industrial catalytic system is its complexity which can be represented by three main, interwoven factors:
(a) multi-component catalyst composition;
(b) complexity of the catalytic reaction itself; and
(c) temporal change of the catalyst composition under the influence of the reaction medium.
The Temporal Analysis of Products (TAP) reactor system has been developed to examine the detailed adsorption/desorption and surface reaction behaviour of heterogeneous catalysts in the gas phase1,2 and has been applied to a wide range of reactions and catalysts.1–6 The TAP reactor operates on the premise of injecting a small gas pulse of very short duration into an evacuated micro-reactor containing a packed bed of particles. The duration of the inlet pulse is in the order of 500 µs, which provides the technique with a sub-millisecond time resolution. When the number of molecules in the pulse is sufficiently small (∼10−10 moles), the convective flow disappears and gas transport occurs by Knudsen diffusion. Due to the low inlet pulse intensity, the pulse-response TAP experiment provides an opportunity to control the catalyst composition.2 In a single-pulse TAP experiment, the changes in the catalyst composition during the experiment are very small compared with the amount of surface active sites because of the small pulse intensity. In a multi-pulse TAP experiment, the catalyst composition can be changed gradually, in a well-controlled manner.
Mass spectrometry has been extensively used as a tool of choice in the investigation of both un-catalyzed and catalyzed chemical reaction mechanisms due essentially to its high sensitivity and time resolution.1,7–16 These characteristics make mass spectrometers (MS) ideal for analyzing reactant consumption, product formation and intermediate identification,8 which can be combined to elucidate mechanisms which cannot be achieved by any other instrument.11 A number of different types of mass spectrometer have been utilised for this purpose such as ion trap devices (Paul traps, Fourier transform ion cyclotron resonance MS) and transmission devices (quadrupole, sector and time of flight MS).9Ion trap MS are limited in terms of the numbers of ions that can be held (100–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and there is a greater chance of sample decomposition when ion storage is used. This characteristic puts this type of device at a disadvantage with respect to transmission instruments where analysis is carried out directly, without ion storage.9
000) and there is a greater chance of sample decomposition when ion storage is used. This characteristic puts this type of device at a disadvantage with respect to transmission instruments where analysis is carried out directly, without ion storage.9
In the case of the TAP reactor, to date, quadrupole mass spectrometers (QMS) have been used almost exclusively to monitor transient kinetic experiments. These provide a good time resolution, are versatile and have a sufficiently wide dynamic range to provide sufficient sensitivity. Their key drawback is that in order to achieve ms time resolution, the QMS must be locked to one particular atomic mass unit (AMU) and switching between AMUs typically takes several milliseconds. Thus, to observe a number of different AMUs for reactants and products with sub-millisecond time resolution, the corresponding number of transient experiments must be performed.
In the case of TAP technique, this limitation has been partially overcome by Moulijn and co-workers17 using the MULTITRACK modification. In this system four QMSs were installed in a series manner to enable observation of different AMUs simultaneously. However, in this case the overall setup becomes much more complex as each QMS is not geometrically equivalent and, therefore, needs to be modelled differently. Even with this modification, the number of AMUs which can be recorded simultaneously is still only limited to the number of mass spectrometers used.
Time of flight (ToF) mass spectrometers offer the opportunity to observe all masses simultaneously. In this type of mass spectrometer, all ions which have identical kinetic energy are injected in a flight tube where they separated according to their respective masses. This is achieved by the ions with smaller masses arriving at the detector faster than ions with larger masses i.e. ions with smaller mass will have a shorter “flight time”.18 However, until recently, ToF MS were not well suited for observing fast time-dependencies like the ones monitored in a TAP pulse-response experiment. The coupling of ToF mass spectrometers to the TAP reactor has been investigated previously.19 Randall et al. adapted a linear time of flight mass spectrometer to a TAP system20 which provided millisecond time resolution but was not adequate in terms of the sensitivity required for the TAP technique.6 Sakamoto and co-workers at Toyota have also developed a time of flight mass spectrometer for a TAP-like system.21–25 However, from a detailed consideration of the intensity of pulses used, this system may be limited in terms of sensitivity since pulses used in the reported experiments are several orders of magnitude higher than the upper limit of Knudsen diffusion. Under such conditions it is impossible to use traditional TAP-models with a simple linear diffusional transport term and subsequently the TAP theory cannot be applied to data interpretation thus limiting the analysis to only qualitative information.
It is clear that one of the main obstacles in developing a ToF for TAP applications lies with obtaining sufficient sensitivity of the detector at millisecond time resolution. An important additional difficulty with respect to ToF systems is associated with linearity issues of the detection system.26 To be able to quantitatively and accurately analyse the fast TAP pulse-responses, it is necessary to have a high fidelity between the detector response and the time dependent partial pressure being measured. This implies a fully linear detector over the dynamic range explored with suitable time resolution.
In this article, a TAP reactor equipped with a Time of flight (ToF) mass spectrometer suitable in terms of sensitivity, detector response and time resolution, for application in fast transient TAP kinetic catalyst characterization is presented and discussed. The discussion includes the technical difficulties associated with the adaptation of ToF mass spectrometry to TAP application and solutions implemented in terms of this adaptation. The performance of the ToF mass spectrometer, especially the linearity of the specific detector over the full dynamic range was validated using various tests under steady-state and transient pulse-response TAP conditions.
Technical details
TAP system
All experiments were carried out using a TAP-1 reactor system (Fig. 1) which has been described in detail previously.27,28 In summary, the TAP-1 reactor is comprised of three chambers namely a reactor chamber, differential chamber and detection chamber. The reactor chamber is pumped by an oil diffusion pump and houses the stainless steel micro-reactor (41 mm in length and 5.5 mm i.d.). Two turbo molecular pumps (Pfeiffer Vacuum TMU 521 DN) maintain the vacuum in the differential and detector chambers. The establishment of the ultra-high vacuum conditions ensures the continuous evacuation of the TAP micro-reactor. A temperature controller (Eurotherm 818 model) and a Type K thermocouple are incorporated into the system. The thermocouple allows the catalyst bed temperature to be monitored. The temperature controller enables the temperature to be set so that reactions can be investigated over a range of temperatures (15–750 °C). The required ratio of reactant gases are mixed in the gas mixing manifold prior to pulsing into the micro-reactor. It is also possible to vary the pulse intensity using pressure regulators incorporated into the gas mixing manifold. | ||
| Fig. 1 Photograph of the QUB TAP-1 reactor with ToF MS installed. | ||
To validate the ToF-MS for Knudsen diffusion a one zone micro-reactor packing configuration consisting of silicon carbide (particle size 0.5 nm) was used. Helium, neon, nitrogen and argon were pulsed over the inert packing at 17, 50, 100, 150, 200, 250 and 300 °C. Each pulse response was recorded for 2 s, and the experiments for each probe molecule involved the averaging of 100 pulses for more accurate analysis.
The TAP ToF MS system (shown in Fig. 1 and 2) developed in this work is comprised of four ion optical elements. These are the ion source, the transfer optics, the electrostatic analyser (ESA) and the charged particle buncher (which incorporates the detector). All these major elements of the TAP ToF MS and the orientation of these elements are illustrated in Fig. 2.
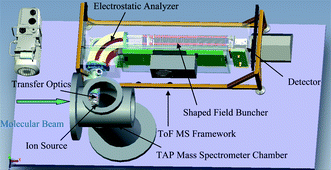 | ||
| Fig. 2 Schematic of the ToF MS framework for the TAP-1 system. | ||
Time of flight mass spectrometer—the ion optical elements
A description of the constitutive parts of the ion optical element is given in the following sections.The transfer optics is formed by an accelerating einzel lens which collects and focuses the ion beam from the source into the plane of a pressure step aperture. This aperture separates the TAP detector chamber from the vacuum environment of the ToF MS analyser. X and Y deflection plates provide steering for the beam in order to maximise transmission through this aperture. Due to the length of the transfer optical column (20 cm) a mumetal shield was fitted around the optical arrangement to guard against attenuation of ions of hydrogen, deuterium and helium by stray magnetic fields.
Fig. S4† depicts a potential energy representation of the passage of the ion beam through the transfer optics. The beam is accelerated from 100 eV to nearly 700 eV by the action of the accelerating einzel lens. It is then focused into the pressure step aperture.
Ions enter the buncher continuously from the left at low energy (shown in black in Fig. S6†). As the operation of the buncher commences, the potential of the ions increases which then accelerate out of the buncher towards the detector. The time markers show the ions coming into time focus near the detector. The potential is shown in blue in Fig. S6† where the peak heights are proportional to the potential.
It takes an ion of mass to charge ratio (m/z) of 100 approximately 80 µs to traverse the 30 cm along the axis of the 30 co-axial electrodes which form the shaped field buncher. During this time the potential on all of the electrodes is held very close to 0 V, presenting no field to the advancing ion beam.
The bunching action is activated every 100 µs by applying approximately 7 kV to the first electrode of the buncher. Successively lower voltages appear on the remaining electrodes, creating the shaped field. This field is applied within a time of 20 ns and is maintained for a period of 20 µs, after which all charged particles have exited the buncher and been directed towards the detector. If the shaped field is correct then every ion of a given charge to mass ratio will arrive at the detector simultaneously, irrespective of its position within the buncher when the buncher commences operation. The field then returns to zero for the remaining 80 µs, allowing the charged particle beam to re-fill the buncher until the cycle is repeated.
Results and discussion
ToF MS detector design modifications
Initially, a discrete dynode electron multiplier detector was employed to collect the signal. This detector had a very fast pulse width of less than 2 ns but it used a series of capacitors in parallel with the resistors on the penultimate dynodes (RC circuits) in order to maintain the gain in a ToF experiment, where the current drawn from the last dynode can exceed 10 mA. Unfortunately, the time constants of these RC circuits were in the order of ∼100 ms which caused the gain of the detector to vary on a timescale which coincided with the timescale of interest for a TAP experiment. The use of this detector led to the presence of an undulating wave superimposed upon the true trace. In order to circumvent this issue, a hybrid detector design comprising a single micro-channel plate and a photomultiplier was employed. Although the photo-multiplier is a discreet dynode device, similar to the original detector employed, it sustains the voltages on the dynodes with a current pulse every 10 µs from a Cockcroft–Walton supply. In addition, no capacitor–resistor networks (RC circuits) were used eliminating the variation in gain.Linearity of the detector
The linearity of the detector can also be affected by the statistical variation of the number of ions that are detected during the recording of a TAP pulse response. If there is a low ion event count rate, i.e. only a small number of single ions reaching the detector, it is possible to employ a pulse counting system. A pulse counting system amplifies the signal without compromising the integrity of the signal. For example if it is assumed that the maximum signal intensity corresponds to the detection of one ion in each mass spectrometer cycle (100 µs), the reaction trace is set at 10 ions with duration of 1 ms. Supposing each TAP pulse response is the sum of 100 reaction traces, the total number of ions contributing to a TAP pulse response is 1000. In this instance the signal to noise ratio is ∼31.6. In order to preserve the linearity of the detector it is necessary that all ion events are taken into account. This must range from single ion events in the ion counting region to the multiple ion events of the proportional multiple ion counting region. However, saturation of a pulse counting system occurs rapidly when multiple ion events are detected and so it is not possible to preserve the linearity of the detector with this method. During the cycle of the mass spectrometer, simultaneous multiple ion events must be recorded with a high degree of accuracy. This accuracy is required to ensure signal intensity is proportional to the actual molecular beam exit flow from the micro-reactor. In order to achieve this, a high gain electronic amplifier coupled with a fast digitizer consisting of multiple digital levels was employed to ensure detector linearity during simultaneous multiple ion events.Fig. 3 reports a typical example of an area normalized argon response (shown in blue) recorded by the ToF MS and the best theoretical fit, shown in red (see ESI† for theoretical details). The difference between the experimental and the theoretical curves is plotted in green and clearly shows a significant discrepancy between experimentally observed and theoretical responses. A number of features are discernible. The experimental curve had a slower ascent prior to the peak compared with that of the theoretical curve, which produced an initial negative difference between these curves. After the response maximum, the experimental curve is found to decrease faster than the theoretical curve, which again resulted in a negative difference between the curves.
 | ||
Fig. 3 Comparison of the area-normalized experimental curve (■) and theoretical model (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and difference between traces (×) for argon, monitored at 40 AMU, pulsed over SiC at 100 °C. ) and difference between traces (×) for argon, monitored at 40 AMU, pulsed over SiC at 100 °C. | ||
The observed discrepancy between ToF recording and TAP theory suggested that the detection sensitivity increased with the signal and caused stretching of the response possibly due to different efficiencies when detecting single compared with multiple ions. Such variations have been reported by Hunter and Stresau26 wherein it was thought that ToF MS detection systems have three regions of detection efficiency, as shown in Fig. 4. Region 1 represents the detection efficiency when single ions reach the detector per cycle, which provides lower detection efficiency. Region 3 represents the detection efficiency when groups of thousands of ions arrive to the detector per cycle, which corresponds to higher detection efficiency. Region 2 represents intermediate detection efficiency and this occurs when single ions as well as groups of thousands of ions arrive at the detector per cycle.
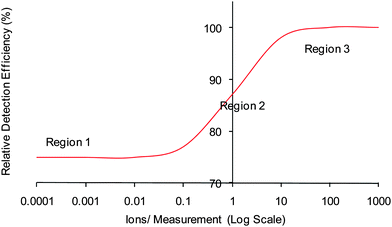 | ||
| Fig. 4 Schematic of the relative detection efficiency found in a ToF MS, as reported by Hunter and Stresau.26 | ||
Hunter and Stresau26 related this inconsistency in the detection efficiency to the threshold level of the analogue-to-digital converter (ADC) and introduced a calibration function in order to linearise the relationship. This function relates the measured ion abundance to the actual number of ions that reach the detector per measurement cycle. The function K(Iab) is defined in eqn (1):
 | (1) |
To verify the hypothesis that the problem was linked to the detector behaviour and to evaluate the K-function for the specific system employed in this study, neon (20 and 22 AMU) and residual gas (18 and 28 AMU) were used to calibrate the ToF MS with a QMS. The total pressure of the system was recorded separately in order to confirm that the sum of all the partial pressures recorded with the QMS could be related to the actual pressure of the vacuum chamber. The calibration function, K (eqn (1), K-function = number of neon ions observed by ToF/partial pressure of neon observed by QMS × normalization factor), plotted against the logarithm of the number of neon ions observed in the ToF signal, as illustrated in Fig. 5. The results indicated that the K-function significantly increases as the partial pressure of neon increases, exhibiting a trend similar to that reported by Hunter and Stresau.26
 | ||
| Fig. 5 K-Function (number of neon ions observed by ToF/partial pressure of neon observed by QMS × normalization factor) vs. logarithm of the number of neon ions observed in the ToF signal with (○) increasing neon partial pressure and (▲) decreasing neon partial pressure. | ||
To resolve the problems modifications to the hardware of the ToF MS were made. In particular, the values for the gain of the detection system and the DC offset of the transient digitiser were adjusted to make the relative detection efficiencies in both the low count rate and the high count rate regimes equal. Comparison between the QMS and ToF-MS showed a constant ratio of ToF intensity/QMS intensity with increasing pressure of neon (Fig. 6).
 | ||
| Fig. 6 K-Function (number of neon ions observed by ToF/partial pressure of neon observed by QMS × normalization factor) vs. logarithm of the number of neon ions observed in the ToF signal, after modifications to the ToF MS detector systems with (○) increasing neon partial pressure and (▲) decreasing neon partial pressure. | ||
Data acquisition
A typical TAP experiment requires an actuation signal to inject a pulse of gas mixture into the micro-reactor and simultaneous signals to initiate the acquisition electronics and start the buncher operation. Fig. 7 shows a timing diagram for a TAP ToF MS experiment initiated at 0 s. This is a typical sequence for which it is desired that the temporal resolution of the first dataset transferred to disc is 1 ms. In this case, the microsecond timescale illustrates the time taken for data collection compared with the time to fill the buncher with ions. The millisecond timescale illustrates 10 buncher fire data acquisition sets, which are recorded after the buncher is instructed to fire and are summed to give a mass spectra from 1 AMU to 50 AMU (0–1 ms shown in blue, 1–2 ms shown in red and 2–3 ms shown in green in Fig. 7). The second timescale shows an imagined temporal response of a particular mass channel, where the first data point represents the sum of ten individual spectra (therefore representing 1 ms of data). The second data point represents the sum of the next ten individual spectra, and so on until 30 ms of the reaction trace for one particular mass channel is represented.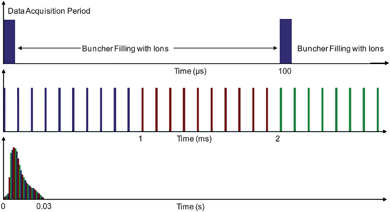 | ||
| Fig. 7 A timing diagram for the three timescales of the evolution of a TAP experiment. The microsecond timescale illustrates the time taken for data collection compared with the time to fill the buncher with ions. The millisecond timescale illustrates 10 buncher fire data acquisition sets, which are summed to give mass spectra from 1 AMU to 50 AMU (0–1 ms shown in blue, 1–2 ms shown in red and 2–3 ms shown in green). The second timescale shows an imagined temporal response of a particular mass channel, where the first data point represents the sum of ten individual spectra (therefore representing 1 ms of data). The second data point represents the sum of the next ten individual spectra, and so on until 30 ms of the reaction trace for one particular mass channel is represented. | ||
The detector operates with rapid rise and fall time of ∼2 ns. An 8 bit digital copy of the signal from the detector is stored in a transient digitizer (temporal resolution = 1 ns) for a period of 4 µs after the buncher fires. Such spectra can be summed by a factor selectable by software from 2, 5, 10 and so on, yielding temporal resolutions of the instrument of 200, 500, and 1000 µs with a corresponding improvement in signal to noise ratio.
A spectrum length of 4 µs gives the instrument a mass range of ∼1 to 50 AMU. A greater mass range can also be incorporated by digitizing with a resolution of 2 ns. This gives a range of up to 200 AMU. The spectral data are stored as a series of four thousand 16 bit words and these data are transferred to disc and stored at up to 40 MB s−1. This limit is incurred as a consequence of the bus structure which operates via a Peripheral Compute Interface (PCI). This solution was employed to reduce the volume of data to be processed, so that the analytical capability of the instrument would not be compromised.
Validation of TAP ToF MS
The typical procedure to validate that the TAP system is functioning correctly is to verify the agreement between the observed diffusion responses and TAP Knudsen diffusion theory. In this case, the TAP micro-reactor is filled with inert particles (SiC, quartz, etc.) to reduce any adsorption processes then responses for a range of probe molecules with different masses are recorded as a function of the pulse intensity and temperature. These are compared with the response shapes derived from Knudsen Diffusion TAP Theory (see ESI†). It should be noted that the performance of the TAP system employed has been validated using the QMS prior to the ToF MS installation.17,18Fig. 8–11 show the comparison between theoretical curves and typical responses observed for argon, nitrogen, neon and helium. In all cases the agreement between theory and experiment is excellent. The argon responses from single ionized (40 AMU) and double ionized atoms (20 AMU) closely follow each other.
 | ||
| Fig. 8 Comparison of theoretical curves and experimental argon responses observed at AMU = 20 and AMU = 40 at 50 °C ((×) AMU40, (△) AMU 20, (blue line) theory) and at 300 °C ((□) AMU 40, (○) AMU 20, (black line) theory). | ||
 | ||
| Fig. 9 Comparison of theoretical curves and experimental nitrogen responses observed at AMU = 14 and AMU = 28 at 17 °C ((×) AMU 28, (△) AMU 14 (blue line) theory) and at 250 °C ((□) AMU 28, (○) AMU 14, (black line) theory). | ||
 | ||
| Fig. 10 Comparison of theoretical curves and experimental neon responses observed at 50 °C ((×) AMU20, (blue line) theory) and at 300 °C ((□) AMU 20, (black line) theory). | ||
 | ||
| Fig. 11 Comparison of theoretical curves and experimental helium responses observed at 50 °C ((×) AMU4, (blue line) theory) and at 300 °C ((□) AMU 4, (black line) theory). | ||
In addition to the comparison between the observed and theoretical responses, the 4th criterion (which describes the dependence of Knudsen diffusivity on temperature and molecular weight, see eqn (S8) in ESI†) was also evaluated. The residence times (first moment) for all responses (recorded over the same micro-reactor packing) were calculated and plotted vs. (molecular weight/temperature in K)1/2 (Fig. 12). It is clear that all the data points followed a single straight line as expected from the theory.
 | ||
| Fig. 12 Residence time (first moment, M1) plotted vs. (molecular weight/temperature (K))1/2 for (△) helium, (◇) neon, (●) nitrogen and (■) argon. | ||
Fig. 13 displays six pulse response curves of argon which were recorded at 16 °C using similar pulse intensities. The six experiments were carried out at regular intervals during a 30 h period. The results clearly show the reproducibility over the period of the six experiments was excellent demonstrating the high degree of stability of the system.
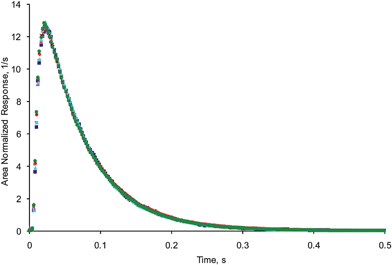 | ||
| Fig. 13 Comparison of traces on pulsing argon over SiC at 16 °C, using the ToF-MS after (■) 0 h, () 3 h, (◆) 6 h, (▲) 24 h, (×) 27 h and (●) 30 h. | ||
As already described, one of the major advantages of a ToF MS is the possibility to analyse multiple masses simultaneously at a high time resolution. This provides the opportunity of analysis of complex mixtures of reactant and products and, most importantly, short lived intermediates. However, to achieve this aim it is necessary to ensure the correct deconvolution of specific responses from the combined fragmentation patterns. In order to verify that this could be achieved, the fragmentation patterns of ethane, propane and butane were measured separately by pulsing a 50/50 mixture of each gas in argon followed by a mixture of the gases with argon (20/35/25/20, argon/butane/propane/ethane) over SiC. To deconvolute the data 14 AMUs were followed: 58, 57, 50, 44, 43, 42, 41, 39, 30, 29, 28, 27, 26, and 15 (a mass spectrum typical of the 20/35/25/20 mix of argon/butane/propane/ethane investigated is reported in Fig. S7†). The fragmentation patterns of butane, propane, ethane and gas mix were recorded as column vectors ![[X with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0058_20d1.gif) Bt, Pr, Et and mix, respectively. Each vector consisted of 14 elements. Each element is equal to the ratio of HC intensity observed at specific AMU (zeroth moment MAMU,0) to the intensity of argon observed at AMU = 40 (zeroth moment MAr40,0) in the same experiment with the 50/50 mixture pulsed, as is expressed in eqn (2):
Bt, Pr, Et and mix, respectively. Each vector consisted of 14 elements. Each element is equal to the ratio of HC intensity observed at specific AMU (zeroth moment MAMU,0) to the intensity of argon observed at AMU = 40 (zeroth moment MAr40,0) in the same experiment with the 50/50 mixture pulsed, as is expressed in eqn (2):
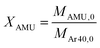 | (2) |
The following fragmentation analysis was performed. From superposition principle, the raw observed responses at each AMU are the sum of the contributions from all three hydrocarbon molecules as expressed in eqn (3):
 | (3) |
![[F with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0046_20d1.gif) raw(t) are vectors with 14 elements where each element is the raw (unnormalized) response observed at specific AMU. After normalization to the argon intensity at AMU = 40, eqn (3) can be modified to the form shown in eqn (4):
raw(t) are vectors with 14 elements where each element is the raw (unnormalized) response observed at specific AMU. After normalization to the argon intensity at AMU = 40, eqn (3) can be modified to the form shown in eqn (4): | (4) |
n(t) are also vectors with 14 elements where each element is the response at a specific AMU normalized to the argon intensity at AMU = 40 assuming a 50/50 mix of HC/Ar. The coefficients nBt, nPr and nEt determine the composition of the hydrocarbon mix. Assuming no conversion in the reactor packed with the inert particles (eqn (5)):| nBt + nPr + nEt = 1 | (5) |
For each hydrocarbon molecule the responses observed at different AMUs should have exactly the same shape but should vary in terms of intensity following the corresponding fragmentation pattern. The fragmentation vectors Bt, Pr, and Et by definition determine these intensity distributions. Thus, the response vector for each hydrocarbon can be presented as shown in eqn (6):
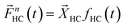 | (6) |
| n(t) = nBtBtfBt(t) + nPrPrfPr(t) + nEtEtfEt(t) | (7) |
In eqn (7), the fragmentation vectors, composition coefficients, and response shape functions are explicitly distinguished. In a matrix form, it can be presented as shown in eqn (8):
 | (8) |
![[X with combining circumflex]](https://www.rsc.org/images/entities/i_char_0058_0302.gif) and composition coefficients multiplied times area-normalized responses forms the response vector n
and composition coefficients multiplied times area-normalized responses forms the response vector n![[f with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0066_20d1.gif) . This system of linear equations is over-determined for the response vector n (3 elements versus 14 AMUs). It can be solved using vector algebra method that minimizes squared displacement, as demonstrated by eqn (9):
. This system of linear equations is over-determined for the response vector n (3 elements versus 14 AMUs). It can be solved using vector algebra method that minimizes squared displacement, as demonstrated by eqn (9):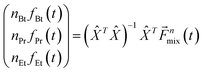 | (9) |
Thus, the responses for each hydrocarbon molecule weighted with composition factors can be determined from the mixture responses observed at different AMUs and fragmentation matrix, . If eqn (9) is integrated with respect to time, area-normalized functions f(t) become equal to one and the response vector  turns into fragmentation vector of the mixture mix, as represented in eqn (10):
turns into fragmentation vector of the mixture mix, as represented in eqn (10):
 | (10) |
Eqn (10) was used to determine the composition of the hydrocarbon mixture. The comparison of the calculated composition with the prepared composition is given in Table 1 and shows excellent agreement.
In a second step, the area normalized responses for butane, propane, and ethane were calculated using eqn (9). Fig. 14 and 15 report these responses at 50 and 100 °C, respectively, plotted together with the theoretical fits. The fitting curves were calculated by rescaling the fitting curve of corresponding argon responses based on Knudsen diffusion theory.2 The square root of ratio of molecular weight was used as a rescaling factor in eqn (S5) (see ESI†). The agreement is again very convincing.
 | ||
| Fig. 14 Ethane: (□) experimental, (green line) theory. Propane: (△) experimental, (red line) theory. Butane: (◇) experimental, (blue line) theory. Experimental responses were deconvoluted from the fourteen AMU fragment signals of a hydrocarbon mixture of butane, propane and ethane, pulsed at 50 °C. Theoretical curves were obtained from corresponding argon responses, using the Knudsen diffusion TAP model. | ||
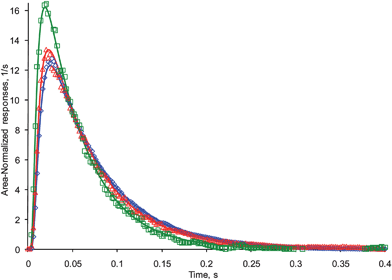 | ||
| Fig. 15 Ethane: (□) experimental, (green line) theory. Propane: (△) experimental, (red line) theory. Butane: (◇) experimental, (blue line) theory. Experimental responses were deconvoluted from the fourteen AMU fragment signals of a hydrocarbon mixture of butane, propane and ethane, pulsed at 100 °C. Theoretical curves were obtained from corresponding argon responses, using the Knudsen diffusion TAP model. | ||
These results prove that the QUB TAP-1 ToF system can be a very useful tool for analysis of complex mixture of different hydrocarbons in fast transient experiments with the following advantages:
(1) Possibility to easily observe the complete fragmentation pattern.
(2) Possibility to identify unexpected products from the observed signal time change.
(3) Wider range of pulse intensity (no need for many pulses to monitor many AMUs).
(4) Increased experimental integrity as all mass channels are collected and stored in the same instant (in a QMS, reactor conditions could change slightly between recording one mass channel and the next).
Conclusions
A time of flight mass spectrometer (ToF MS) which has been customized for adaptation to Temporal Analysis of Products (TAP) kinetic experiments has been developed and validated. It has been demonstrated that this TAP ToF MS has the detector linearity, reproducibility and high sensitivity that is essential to accurately analyze TAP pulse response experiments.The linearity of the detector has been verified and this means that the data collected is a proportional representation of the molecular beam formed from the exit flow of the TAP micro-reactor. The stability and reproducibility of the instrument have been demonstrated via the experiment which was repeated at intervals during a 30 h period, which obtained almost identical results each time. The sensitivity is such that good signal to noise ratio can be observed with low pulse intensities, thereby, ensuring the operation of any experiment within the Knudsen diffusion regime. An excellent agreement has been found between experimentally observed responses and theoretical Knudsen diffusion responses. Furthermore, it has been demonstrated that the QUB TAP ToF may be employed for the analysis of complex mixtures of different hydrocarbons in fast transient experiments. This attribute has been verified by highly accurate identification of the mixture composition and by the deconvolution of the responses of each hydrocarbon in the mixture, where excellent agreement was found with Knudsen diffusion theory.
The successful development of the TAP ToF MS is expected to stimulate further application of modified ToF mass spectrometers to other fast transient techniques.
Acknowledgements
We would like to thank the EPSRC for funding as part of the Carmac and CASTech projects and DEL for studentship funding (KM and NM).References
- J. T. Gleaves, J. R. Ebner and T. C. Kuechler, Catal. Rev. Sci. Eng., 1988, 30, 49 CAS.
- J. T. Gleaves, G. S. Yablonskii, P. Phanawadee and Y. Schuurman, Appl. Catal., A, 1997, 160, 55 CrossRef CAS.
- O. P. Keipert and M. Baerns, Chem. Eng. Sci., 1998, 53, 3623 CrossRef CAS.
- M. Olea, M. Kunitake, T. Shido, K. Asakura and Y. Iwasawa, Bull. Chem. Soc. Jpn., 2001, 74, 255 CrossRef CAS.
- G. S. Yablonsky, M. Olea and G. B. Marin, J. Catal., 2003, 216, 120 CrossRef CAS.
- J. Pérez-Ramírez and E. V. Kondratenko, Catal. Today, 2007, 121, 160 CrossRef CAS.
- S. L. Shannon and J. G. Goodwin, Jr, Chem. Rev., 1995, 95, 677 CrossRef CAS.
- S. M. Thornberg and D. Hunka, Dekker Encycl. Nanosci. Nanotechnol., 2004, 1, 1381 Search PubMed.
- P. Chen, Angew. Chem., Int. Ed., 2003, 42, 2832 CrossRef CAS.
- D. Fabris, Mass Spectrom. Rev., 2005, 24, 30 CrossRef CAS.
- S. Gronert, Chem. Rev., 2001, 101, 329 CrossRef CAS.
- C. Market and A. Pfaltz, Angew. Chem., Int. Ed., 2004, 43, 2498 CrossRef.
- C. Market, P. Rösel and A. Pfaltz, J. Am. Chem. Soc., 2008, 130, 3234 CrossRef CAS.
- C. A. Müller and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 3363 CrossRef.
- C. A. Müller, C. Market, A. M. Teichert and A. Pfaltz, Chem. Commun., 2009, 1607 RSC.
- A. M. Teichert and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 3360 CrossRef CAS.
- T. A. Nijhuis, L. J. P. van den Broeke, M. J. G. Linders, M. Makkee, F. Kapteijn and J. A. Moulijn, Catal. Today, 1999, 53, 189 CrossRef CAS.
- B. A. Mamyrin, Int. J. Mass Spectrom., 2001, 206, 251 Search PubMed.
- J. T. Gleaves, G. Yablonsky, X. Zheng, R. Fushimi and P. L. Mills, J. Mol. Catal. A: Chem., 2010, 315, 108 CrossRef CAS.
- H. T. Randall, P. L. Mills and J. S. McCracken, Stud. Surf. Sci. Catal., 1999, 122, 209 CAS.
- T. Motohiro, Y. Kizaki, Y. Sakamoto, K. Higuchi, Y. Watanabe and S. Noda, Appl. Surf. Sci., 1997, 121/122, 319 CrossRef.
- T. Motohiro, Y. Kizaki, Y. Sakamoto, K. Higuchi, T. Tanabe, N. Takahashi, K. Yokota, H. Doi, M. Sugiura and S. Noda, Appl. Surf. Sci., 1997, 121/122, 323 CrossRef.
- K. Okumura, Y. Sakamoto, T. Kayama, Y. Kizaki, H. Shinjoh and T. Motohiro, Rev. Sci. Instrum., 2007, 78, 104102 CrossRef.
- Y. Sakamoto, K. Kizaki, T. Motohiro, Y. Yokota, H. Sobukawa, M. Uenishi, H. Tanaka and M. Sugiura, J. Catal., 2002, 211, 157 CAS.
- Y. Sakamoto, T. Motohiro, S. Matsunaga, K. Okumura, T. Kayama, K. Yamazaki, T. Tanaka, Y. Kizaki, N. Takahashi and H. Shinjoh, Catal. Today, 2007, 121, 217 CrossRef CAS.
- K. L. Hunter and R. W. Stresau, Proceedings of the 46th ASMS Conference on Mass Spectroscopy and Allied Topics, Orlando, Florida, 1998.
- A. Goguet, S. O. Shekhtman, R. Burch, C. Hardacre, F. C. Meunier and G. S. Yablonsky, J. Catal., 2006, 237, 102 CrossRef CAS.
- S. O. Shekhtman, A. Goguet, R. Burch, C. Hardacre and N. Maguire, J. Catal., 2008, 253, 303 CrossRef CAS.
-
V. C. Parr, S. P. Thompson, M. D. Mills, Charged Particle Buncher, US Pat., 7045792, 2006.
Footnote |
| † Electronic supplementary information (ESI) available: schematic drawings of the instrument, spectrum of a hydrocarbon mix in the 0–60 AMU range, details of the Knudsen diffusion TAP theory and definitions of notation used. See DOI: 10.1039/c0an00435a |
| This journal is © The Royal Society of Chemistry 2011 |