Mediator-free amperometric glucose biosensor based on glucose oxidase entrapped in poly(vinyl alcohol) matrix†
Maria Rachele
Guascito
*,
Daniela
Chirizzi
,
Cosimino
Malitesta
and
Elisabetta
Mazzotta
Dipartimento di Scienza dei Materiali, Università del Salento, via Arnesano, 73100, Lecce, Italy. E-mail: maria.rachele.guascito@unisalento.it; Fax: +0039 0832 297100; Tel: +0039 0832 297075
First published on 29th September 2010
Abstract
A simple and novel amperometric biosensor for glucose detection is proposed. It is based on the immobilization of glucose oxidase (GOx) in a poly(vinyl alcohol) (PVA) matrix directly drop casted on a platinum electrode surface (Pt/GOx-PVA). Glucose was determined in the absence of a mediator used to transfer electrons between the electrode and the enzyme. The correlation between peak current (ip) and scan rate has been verified and the effect of pH solution has been checked. Glucose detection has been performed amperometrically at −400 mV by using pulsed amperometric detection (PAD). Under the selected optimal conditions, the biosensor showed low detection limit (10 μM), wide dynamic range (0.1–37 mM) and high sensitivity. The biosensor amperometric response revealed it to be specific to glucose without significant interference from other sugars and electroactive species coexisting with glucose in biological fluids. Response stability was another interesting feature of the developed system as it was almost completely recovered when the biosensor was left in opportune storage conditions (i.e., a response decrease of only 13% after 35 days in air at room temperature). Finally, X-Ray Photoelectron Spectroscopy (XPS) characterization revealed a homogeneous film deposited on the Pt substrate whose structure is also preserved under operative conditions.
1. Introduction
Amperometric enzyme-based biosensors continue to generate much interest in clinical, environmental and quality control analysis. Although the performance of enzyme electrodes is strongly dependent on several factors1 (i.e. the immobilization techniques, thickness and stability of the entrapping membrane, the activity and stability of the entrapped enzyme), they offer several advantages such as the minimum pretreatment of the analyzed matrix and the small sample volume required; moreover, the immobilization helps in forming the required close proximity between the biomaterial and the transducer.2Particular attention has been directed to the realization of amperometric sensors for the clinically significant substrate glucose.3,4 Most methods of glucose detection are based on the use of enzymatic biosensors in which an electron transfer mediator is applied to shuttle the electrons between the redox centers of the enzyme and the electrode.3,5Redox enzymes in fact hardly communicate electrically with conventional electrodes as a thick insulating protein shell surrounds their active center.6,7 For this purpose, natural8–11 and artificial12–15 mediators have been proposed for the development of the so-called ‘first-’ and ‘second-generation biosensors’, respectively.
On the contrary, in mediator-free enzyme biosensors – the ‘third-generation biosensors’ – the electron is transferred directly from glucose oxidase (GOx) to the electrodevia the active site of the enzyme. The main advantage offered by this approach is the possibility of working at a low operating potential – close to that of the redox potential of the enzyme – thus leading to a very high selectivity as the interference of other electroactive species is drastically reduced.5,16,17
Widely used materials for the development of this kind of biosensor are conducting organic salts such as TTF-TCNQ with GOx adsorbed on the crystal surface.6,18,19 Other approaches were based on the direct GOx entrapment on an oxidized boron-doped diamond (BDD) electrode20 and into conducting polymers which were incorporated into the pores of a filtration membrane.21 The design of mediatorless glucose biosensors was performed also by immobilizing the enzyme on the surface of nanomaterial-based electrodes that can enhance the electron transfer kinetics of the enzyme, as for example, carbon nanotubes,22–24 C/Fe nanoparticles composite paste electrode (CFNPE)25 and Si electrodes modified with nanostructured TiO2.16 Recently also polyaniline nanofibers26 and colloidal laponite nanoparticles27 have been successfully applied to mediatorless glucose amperometric detection. The above reported examples show the variety of the proposed approaches for the development of third-generation glucose biosensors.
Nevertheless, the proposed strategies are often time-consuming and labour-intensive as they aim for the development of systems in which the electron transfer distance between the active sites of the enzyme and the electrode is minimized.28 In addition only a few works provide the level of proof for such mediatorless detection.4
In this work, a novel, mediator-free glucose biosensor is proposed. It is fabricated by a simple immobilization procedure of GOx into a biocompatible polymer, poly(vinyl alcohol) (PVA), deposited on a Pt electrode surface (Pt/GOx-PVA). The choice of PVA relies in its recognized properties as an ideal enzyme immobilization material mimicking the enzyme's natural environment and able to stabilize the activity of enzymes, particularly in the case of GOx.29
The very low operating potential (−400 mV vs.SCE) as well as the successful glucose detection in the absence of oxygen evidence that the mediatorless electron transport is occurring. The developed biosensor, prepared by an extremely rapid and simple scheme, revealed a very satisfactory performance in terms of selectivity, stability and reproducibility.
2. Experimental
2.1 Chemicals
GOx (type VII from Aspergillus niger; 179![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units/g), β-D-glucose, D-(−)-fructose, D-sorbitol, uric acid and ascorbic acid were obtained from Sigma. Na2HPO4, NaH2PO4, poly(vinyl alcohol) film (PVA, product number Z300381) were purchased from Aldrich. Flavin adenine dinucleotide (FAD) disodium salt (M = 829.5) was obtained from Sigma. All reagents were analytical grade. Ultrapure water (Millipore Milli-Q, 18.2 MΩcm−1) was used. 1 M stock glucose solutions (prepared every 2 weeks in a phosphate buffer solution (PBS) pH 7.0, I = 0.2) were allowed to mutarotate at room temperature overnight before use.
000 units/g), β-D-glucose, D-(−)-fructose, D-sorbitol, uric acid and ascorbic acid were obtained from Sigma. Na2HPO4, NaH2PO4, poly(vinyl alcohol) film (PVA, product number Z300381) were purchased from Aldrich. Flavin adenine dinucleotide (FAD) disodium salt (M = 829.5) was obtained from Sigma. All reagents were analytical grade. Ultrapure water (Millipore Milli-Q, 18.2 MΩcm−1) was used. 1 M stock glucose solutions (prepared every 2 weeks in a phosphate buffer solution (PBS) pH 7.0, I = 0.2) were allowed to mutarotate at room temperature overnight before use.
2.2 Electrochemical apparatus
All electrochemical experiments were performed on a PalmSens electrochemical workstation (PalmSens Instruments BV, The Netherlands), controlled by computer, using a conventional three-electrode cell with a Pt disk (surface area, 0.0314 cm2) as working electrode, a Pt wire as counter and a saturated calomel electrode (SCE) as reference. All electrochemical experiments were carried out in PBS (pH 7.0, I = 0.2). The buffer was purged with high-purity nitrogen for at least 30 min prior to each electrochemical measurement and a nitrogen environment was kept over the solution to prevent access to the solution from oxygen.2.3 Preparation of the Pt/GOx-PVA biosensor
Before film deposition, the electrode surface was polished with diamond paper, washed and electrochemically pre-treated by cyclic voltammetry (CV) between −200 and +1200 mV vs.SCE in 0.5 M H2SO4 until a steady-state was reached.GOx-modified electrodes (denoted as Pt/GOx-PVA) were prepared by drop casting depositiong directly on the electrode surface a drop of modifier solution, prepared as follows. Firstly, 0.3, 3 and 6 mg of GOx (50, 500 and 1000 units/mL, respectively) were dissolved in 1 mL aqueous solution of 10% PVA in an ultrasonic bath for 5 min. The final GOx concentration (500 units/mL) was selected by comparing the electroactivity of three Pt/GOx-PVA electrodes by CV in the presence of glucose 0.1–30 mM (in the experimental conditions described in Section 2.5). All reported measurements are referred to electrodes prepared with this enzyme amount. The mixture was kept at room temperature for 6 h and then stored at −18 °C for 48 h. It was observed that it was necessary to keep the containing enzyme solution in such conditions to obtain an enhanced biosensor response. This behaviour was in agreement with literature data30 reporting that the subjection of aqueous PVA solutions to freezing–thawing treatment led to reinforced gels owing to the densification of the macromolecular structure. Afterwards 2 μL of GOx-PVA solution were deposited by casting on the Pt surface and left to evaporate for 45 min at room temperature. The calculated total amount of GOx immobilized in PVA film ΓGOx (nmol cm−2) was estimated to be 0.60 nmol cm−2.
Also 2 μL of PVA solution and of FAD 0.03 mg/mL in PVA were deposited in the same way on the Pt electrode to prepare the Pt/PVA and Pt/FAD-PVA electrodes, respectively. In particular, the latter one used in CV experiments was prepared by depositing FAD 0.3 mg/mL in PVA on the Pt electrode to enhance relevant current signals.
2.4 UV-vis spectroscopy measurements
UV-vis measurements were carried out with a Cary 50 spectrophotometer (Varian). The UV-vis spectra of GOx (500 units/mL) both in 10% aqueous solution PVA and in PBS (pH 7.0, I = 0.2) were acquired. 200 μL of GOx-PVA mixture used for biosensor preparation was deposited by casting on a quartz slide and left evaporate for 1 h at room temperature. The UV-vis spectrum was then acquired. For a comparison, UV-vis spectra of FAD solutions (0.03 mg/mL) in 10% aqueous solution PVA and in PBS (pH 7.0, I = 0.2) were analyzed. The same scheme used for GOx-PVA film preparation was used for the deposition of a FAD-PVA film on a quartz slide that was then characterized by UV-vis spectroscopy.2.5 Electrochemical measurements
CV measurements were performed in PBS (pH 7.0, I = 0.2) between −630 and +800 mV vs.SCE in the absence of glucose and with glucose in the range 0.125–30 mM with a scan rate of 50 mV/s.Biosensor response to glucose (0.1–37 mM) was analyzed by pulsed amperometric detection (PAD) by applying a potential of +600 mV for 0.2 s and subsequently switching the potential from −400 mV (measurement potential, maintained for 0.4 s) to −630 mV (applied for 0.2 s). PAD measurements were performed also on bare Pt, on Pt/PVA and on Pt/FAD-PVA electrodes for a comparison.
Selectivity studies were performed by analyzing PAD biosensor response to the most common interferences at their physiological level (uric acid 1 mM, ascorbic acid 1 mM). D-Sorbitol and D-(−)-fructose 1 mM were tested to verify sensor specificity to glucose. Interfering substances were tested also on a Pt/FAD-PVA electrode in the same experimental conditions.
Biosensor storage stability was evaluated by analyzing the amperometric response to glucose (0.1–30 mM) after 3, 5, 30 and 35 days following biosensor preparation. In particular, two different storage conditions (i.e., in clean PBS (pH 7.0, I = 0.2) at +4 °C and dry in a closed vial at room temperature) were tested.
2.6 XPS measurements
XPS spectra were obtained using a Leybold LHS10 upgraded by a PHOIBOS 100 ANALYZER/DETECTOR system (SPECS, Berlin, Germany) and equipped with a twin anode (Mg Kα/Al Kα) non-monochromatized source (operating at 200 W). Survey spectra (FRR, B = 30) were collected at 1 eV steps, whereas detailed spectra (FAT, E0 = 50 eV) for C1s, O1s, N1s, Na1s, P2p and Pt4f regions were collected at 0.1 eV step intervals using multiple sweeps to improve the signal to noise ratio. High-resolution spectra were referenced to the C1s main component binding energy of the adventitious hydrocarbon at 285.0 eV. All XPS spectra were recorded at an electron take-off-angle (TOA) of 90° relative to the sample surface, giving the maximal sampling depth (ca. 5–10 nm).31Pt/GOx-PVA films were analyzed soon after their preparation (a), after being subjected to 10 CV cycles between −630 and +800 mV vs.SCE (scan rate 50 mV/s) in clean PBS (b) and in PBS with glucose 10 mM (c). XPS was used to investigate also Pt/GOx-PVA after amperometric measurements in PAD mode (d) performed in the presence of glucose 10 mM for ca. 900 s (corresponding to the time required for sensor calibration). For a comparison, XPS spectra of PVA (e) were also collected. Immediately prior to being loaded into the XPS spectrometer, films were washed in water and were finally blown-dry using a stream of nitrogen. No X-ray induced decomposition of samples was observed during XPS data acquisition, as manifested by changing peak areas or shifting of positions.
3. Results and discussion
3.1 UV-vis spectroscopy characterization
UV-vis characterization was performed to verify the preservation of GOx integrity when it is embedded in the PVA film. For this purpose, GOx spectra were acquired in the range 240–700 nm in PVA and in PBS solutions (Fig. 1a–b, solid curves) and in PVA film (Fig. 1c–d, solid curves).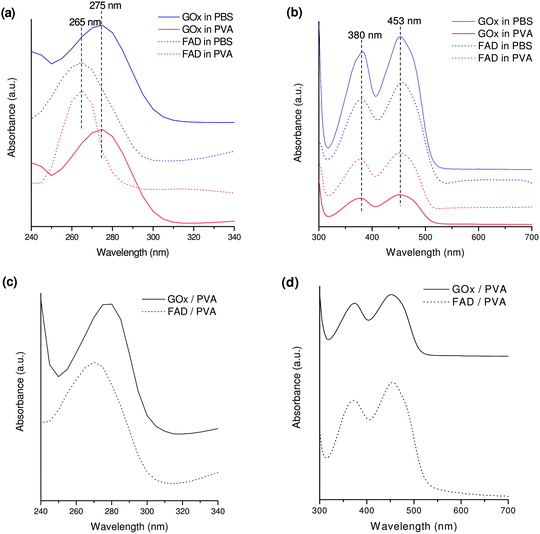 | ||
| Fig. 1 UV-vis spectra for GOx and FAD in PVA and in PBS solutions (a,b) and for GOx-PVA and FAD-PVA films cast on quartz slides (c,d) in different wavelength regions. | ||
Fig. 1a and 1b show GOx characteristic peaks at 275, 380 and 453 nm in PVA and in PBS solutions (red and blue solid curves, respectively). The UV absorption band at 275 nm is assigned to polypeptide chains in protein structure.32,33 The peaks at 380 and 453 nm are characteristics of the oxidized form of the flavin group.32,33 The similarity of GOx spectra in PVA and in PBS solutions suggested that PVA does not determine any modification of the enzyme structure. To confirm this hypothesis, GOx UV-vis spectra were compared with the ones of FAD in the same solutions (Fig. 1a–b, dotted curves). Absorption bands characteristic for FAD34 were observed, located at 265, 380 and 453 nm. The expected differences33,34 between FAD and GOx absorption peaks in the UV region were observed.
Moreover, both in PVA and in PBS solutions the enzyme activity towards glucose oxidation was verified by observing the expected absorption band modifications after glucose addition (1, 5 and 10 mM), due to enzyme reduction (data not reported). The oxidized form of the enzyme was then easily reformed when solutions were saturated by O2.
The preservation of the GOx native structure was verified also in PVA film (Fig. 1c–d, solid curves). It is evident in fact that the position and peak shape of the absorption bands for GOx-PVA film are almost the same as those of pure GOx in PBS. Also in this case, a further verification was provided by the comparison between the spectra of GOx-PVA and FAD-PVA films (Fig. 1c–d, dotted curve) as peak separation of about 10 nm in the UV-vis region was kept constant. It means that the chromophoric groups of FAD responsible for the GOx peaks in the visible region are deeply buried in the enzyme structure27 and thus enzyme structural integrity is perfectly preserved in PVA film. Finally, film exposure to glucose allowed us to verify the preservation of GOx activity in glucose oxidation (data not shown).
The above reported results confirm the great biocompatibility of PVA, already described in the literature.29 PVA is in fact considered an ideal enzyme immobilization material as the abundance of hydroxyl groups provides a microenvironment similar to the enzyme's natural environment and its water content matches that of biological tissue.
3.2 Electrochemical response to glucose on Pt/GOx-PVA by CV
Cyclic voltammetry was used to study the main electrochemical properties of the Pt/GOx-PVA film and to investigate its interaction with glucose. Also Pt and Pt/PVA responses to glucose were evaluated. Results are collected in Fig. 2 showing typical voltammograms recorded respectively on Pt (curve a), Pt/PVA (curve a′) and Pt/GOx-PVA (curve a′′) electrodes in PBS (pH 7.0, I = 0.2). In particular, on Pt/GOx-PVA film (curve a′′) two anodic peaks, at −470 mV (Ia) and −340 mV (IIa), and two cathodic peaks, at −498 mV (Ic) and −357 mV (IIc) were evident. They are located in the region of hydrogen ‘adsorption–desorption’ on Pt35 and could be attributed to hydrogen adsorption–desorption processes typically reported on polycrystalline bare Pt (pc) respectively at Pt (111) and Pt (100) sites.36 These peaks are particularly evident on Pt/GOx-PVA probably because the hydrogen of flavin cofactors FAD/FADH2 is involved in the hydrogen adsorption/desorption process on Pt. The formal potentials, Esurf (I) and Esurf (II), taken by averaging the anodic and cathodic peak potentials for each pair, were −484 mV and −348.5 mV, respectively. | ||
| Fig. 2 CV curves recorded on Pt (a), Pt/PVA (a′) and Pt/GOx-PVA (a′′) in clean PBS (pH 7.0, I = 0.2) and in PBS (pH 7.0, I = 0.2) containing glucose 10 mM (Pt, curve b; Pt/PVA, curve b′; Pt/GOx-PVA, curve b′′). Curves c, c′ and c′′ represent difference voltammograms on Pt, Pt/PVA and Pt/GOx-PVA, respectively. Measurements were performed in nitrogen saturated PBS at a scan rate of 50 mV/s. | ||
Very different shapes of CV curves were recorded on Pt (Fig. 2, curve b), Pt/PVA (Fig. 2, curve b′), and Pt/GOx-PVA (Fig. 2, curve b′′) electrodes after glucose 10 mM addition. The processes related to glucose oxidation on the Pt electrode (Fig. 2, curve b) were evident both in the forward and backward scans according to previously reported data.38 In the oxidation scan, four anodic peaks at −342, −176, +110 and +505 mV were observed. The first peak coincides with hydrogen ‘adsorption–desorption’ on Pt,35 where glucose oxidation is associated with the adsorbed hydrogen atoms.38 Peaks at −176 (the most intense) and +110 mV are related to glucose oxidation in the ‘double layer region’. Finally the peak (very weak) at +505 mV corresponds to the ‘oxide region’.37,38 On the reverse scan there was a substantial oxidation of glucose (peak at −202 mV) and no relevant reduction peaks can be observed in the potential range between −100 and −400 mV (see also curve c).
On the contrary, Pt/PVA electrochemical activity (Fig. 2, curve b′) evidenced only a slight modification after glucose addition (as evidenced also from curve c′) suggesting that the PVA membrane, as expected, reduces the localized active sites on the Pt surface.
An increment of anodic currents was instead recorded on the Pt/GOx-PVA electrode (Fig. 2, curve b′′) on both scans after glucose addition (see also curve c′′). In the oxidation scan, four anodic peaks at −335, −139, +110 and +510 mV were observed. On the reverse scan, there was a high oxidation peak of glucose (at −363 mV) and also in this case no significant reduction signals were observed between −100 and −400 mV.
The role of GOx in enhancing the catalytic response of Pt and Pt/PVA to glucose oxidation was evident especially in the ‘hydrogen region’. A tentative explanation could be that the glucose oxidative process was supported by the redox pair FAD/FADH2 that exchanges electrons directly with the Pt surface. This current increment was so attributed to an irreversible electro-catalytic process of glucose oxidation that probably involves the oxidation of Pt-adsorbed GOx-FADH2. An indirect confirmation of the Pt contribution in the glucose oxidative process was provided by the lack of electrocatalytic activity observed on a glassy carbon electrode in the same experimental conditions (data not shown). Moreover, literature data report the redox potential of the GOx-FAD/GOx-FADH2 pair at −440 mV,4 −418 mV,26 −420 mV39 and −460 mV40 in similar pH values, supporting the hypothesis that the direct electron transfer reaction of GOx plays a key role in the observed electrochemistry. The electro-oxidation of glucose takes place during both direct and reverse scans, as expected for a catalytic process.
Fig. 3 shows Pt/GOx-PVA CV responses to glucose 0–30 mM, at scan rate of 50 mV/s (Fig. 3b). For a comparison, CV profiles of Pt/PVA in the presence of glucose are also reported (Fig. 3a). Fig. 3b reveals that, on increasing glucose concentration, oxidation peaks in hydrogen adsorption/desorption region increase and collapse to form a single broad peak at −335 mV always accompanied from a broad oxidation peak in the reverse scan at −360 mV. Also the oxidation peak in the double layer region (between −200 and 0 mV) increases in both scans. Pt/GOx-PVA CV responses clearly showed that the oxidation of glucose is an irreversible process strictly related to the hydrogen adsorption–desorption redox process localized on specific Pt sites. It was evidenced that there is a synergistic effect between Pt and GOx in the activation of glucose oxidation, especially in the ‘hydrogen region’, confirming that hydrogen is directly involved in this process.
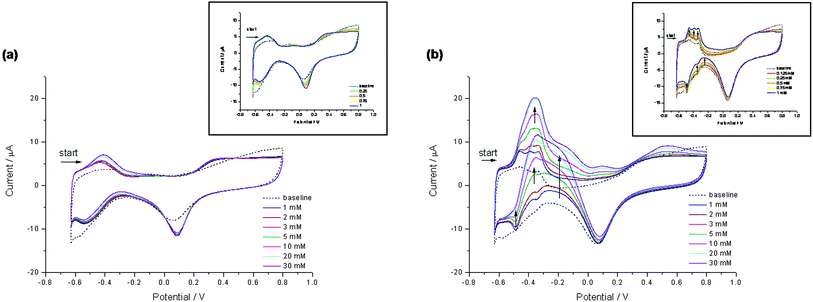 | ||
| Fig. 3 CV curves on Pt/PVA (a) and on Pt/GOx-PVA (b) in the presence of different glucose concentrations in the range 0–30 mM in nitrogen saturated PBS (pH 7.0, I = 0.2). Insets represent CVs obtained in the presence of glucose concentrations lower than 1 mM. Scan rate 50 mV/s. | ||
On the contrary, no significant modification of CV curves was observed on Pt/PVA by increasing glucose concentration (Fig. 3a).
Catalytic current in the direct scan ic,dir (evaluated on Pt/GOx-PVA at Epa = −335 mV after the subtraction of the current in the absence of glucose) was plotted against glucose concentrations between 1 and 30 mM (data not shown). ic,dir increased with glucose concentration reaching a plateau at glucose concentrations higher than 10 mM, as expected for a Michaelis–Menten mechanism. A similar trend was observed also considering the catalytic current at −360 mV in the reverse scan (ic,rev), with ic,dir/ic,rev equal to ≈ 1.
In order to verify that the glucose catalytic oxidation process was due to the reduction of FAD linked to the protein and not to the reduction of free FAD molecules released from the enzyme, FAD electrochemical behavior was analyzed by CV and compared with that of GOx. Comparison between CV profiles of Pt/GOx-PVA and Pt/FAD-PVA electrodes between −630 and −200 mV is reported in Fig. S1 (see ESI†). While Pt/GOx-PVA exhibits the two characteristic couples of peaks, FAD shows a different behaviour with redox peak potentials at −422 and −532 mV and a formal potential Esurf equal to −477 mV, in agreement with literature data.41 The difference value of peak potentials on FAD- and on GOx-modified electrodes confirms that the electroactivity observed on the latter is ascribable to prosthetic FAD groups linked to the protein,27 thus supporting UV-vis data on enzyme structural integrity in the adopted conditions.
To better understand electron transfer processes on Pt/GOx-PVA, CV studies were performed in the absence of glucose at different scan rates between 10 and 500 mV/s. Increasing the scan rate, the redox peak currents in the ‘hydrogen region’ showed a linear dependence on the scan rate, indicating a surface controlled electrode process for both pairs (Fig. S2 in the ESI†).
Moreover, both the anodic and cathodic potential peaks showed a shift with the increase of the scan rate v. Fig. S3 (ESI†) shows the variation of Epa and Epc as a function of log v for Ia/Ic and IIa/IIc peak pairs. In the 25–400 mV/s scan-rate interval a linear dependence was evident for both peaks, as expected for a totally irreversible process.42 According to this model, a graph of Ep = f(log v) yields two straight lines with a slope equal to −2.3RT/αnF and intercept −(2.3RT/αnF)log(αnF/RTks) + Esurf for the cathodic peak (eqn (1)). For the anodic peak the slope is 2.3RT/(1 − α)nF and intercept [2.3RT/(1 − α)nF]log[(1 − α)nF/RTks)] + Esurf (eqn (2)).
| Epc − Esurf = (−2.3RT/αnF)log v − (2.3RT/αnF)log(αnF/RTks) | (1) |
| Epa − Esurf = [2.3RT/(1 − α)nF]log v + [2.3RT/(1 − α)nF]log[(1 − α)nF/RTks] | (2) |
Furthermore, an increase in pH (from 3 to 8) causes a negative shift in Ep (I) and Ep(II) slope of respectively −30 mV/pH for peak pair (I) and −36 mV/pH for peak pair (II) (see Fig. S4 in the ESI†). Both values are about half of the reported value of −60 mV/pH for the redox process of FAD/FADH2 related to a direct electron transfer mechanism involving two protons and two electrons.24 The observed changes of the midpoint potential as a function of the pH are very close to previously reported results.47–49 In order to exploit these ΔE/ΔpH values, a process involving two electrons and one proton can be invoked.45,50 It should be in fact considered that flavins possess three oxidation states:50 the fully oxidized flavoquinone (FAD), the flavosemiquinone radical, in either the anionic reduced (FAD−) or neutral form (FADH) and the two-electron reduced flavohydroquinone, which can exist in either anionic (FADH−) or neutral (FADH2) form. It could be hypothesized that in the adopted experimental conditions the anionic form FADH− is the prevailing form and, thus, the further protonation is not promoted. As a consequence, the whole redox mechanism involves two electrons and one proton for each FAD moiety.45,50 Finally, it should be observed that different experimental values (−43.7 mV/pH,51 −44.5 mV/pH,52 −41 mV/pH53) have been reported for similar systems when used as a third-generation electrode. This indicates that in many cases the mechanism is more complex than the one involving two electrons and two protons, probably due to the local chemical environment and to the occurrence of simultaneous reactions.
The effect of scan rate (5–200 mV/s) on CV responses was evaluated also in the presence of glucose 10 mM after correction from baseline recorded without glucose at each of the tested scan rates. The peak current in oxidation was proportional to the square root of the scan rate (eqn (3)) (Fig. S5a in the ESI†) with a determination coefficient R2 = 0.9955.
| ip (μA) = − 2.77 (μA) + 1.26 [(μA)/(mV/s)1/2] v1/2(mV/s)1/2 | (3) |
Moreover, Ep increases with log v (eqn (4)) (Fig. S5b in the ESI†) (R2 = 0.959)
| Ep (mV) = − 370.4 (mV) + 22.49 (mV) log v | (4) |
These results are in good agreement with a diffusion-controlled reaction scheme. Thus, in the explored scan rate range, the model of heterogeneous catalysis on redox chemically modified electrodes can be applied, with a reversible charge transfer reaction and an irreversible chemical reaction.54,55
A catalytic mechanism for the oxidation of glucose was hypothesized (Scheme 1). The presence of the reduced form FADH2 is promoted by the cathodic applied potential. As reported above, it is in equilibrium with FADH− that is the prevailing form in the adopted experimental conditions. The first step is the dehydrogenation of FADH− (Pt-assisted) to form the FAD−radical anion and H radical adsorbed on Pt, with the simultaneous direct transfer of two electrons to the Pt surface generating fully oxidized flavoquinone FAD and H+. After, FAD reacts chemically with glucose to give gluconic acid and to regenerate FADH−. The first step is kinetically enhanced principally in the specific Pt polycrystalline sites.
 | ||
| Scheme 1 | ||
The proposed mechanism takes account of the role played by the Pt surface in the redox process. Nevertheless, it could be hypothesized that also the entrapping membrane affords a certain contribution in promoting the electron transfer. It could keep the GOx molecules adsorbed at the electrode in a particular order to ensure that the electron transfer between their FAD molecules can be facilitated.
Finally, the effect of O2 on the glucose oxidation process was evaluated by studying the CVs in non-deaerated PBS containing glucose 10 mM (Fig. S6 in the ESI†). It is evident that no relevant electrochemical activity was observed in the ‘hydrogen region’, confirming that O2 is not involved in the electrocatalytic mechanism of glucose oxidation.
A tentative explanation of the observed loss of enzyme activity is based on the O2reduction with the subsequent production of H2O2. It is in fact reported56–58 that H2O2 can deactivate GOx by reacting with amino acid functions and hence leading in the loss of enzyme activity.57
3.3 Amperometric response of the biosensor to glucose
Glucose amperometric detection required the application of multistep potential waveforms to the working electrode, which incorporated cleaning and activation steps along with detection. By performing glucose detection at a constant potential in the range between −500 and +100 mV, no current increase was in fact recorded.The applied PAD consisted of 3 steps. The first, highly positive (i.e., +600 mV for 0.2 s), caused oxide formation. The second was slightly negative (−630 mV for 0.2 s) and removed the oxide layer by reduction to reactivate the electrode. During the third step, current was measured at the applied potential of −400 mV for 0.4 s. Such behaviour supports the hypothesis that the monitored redox processes involve the Pt surface. It is in fact reported59 that Pt suffers from a pronounced poisoning effect by some intermediates and/or products suppressing its intrinsic activity by blocking the surface electrode.
By modifying only the detection potential value (in the range between −600 and +800 mV), the effect of the applied potential was studied and −400 mV was selected as optimal value at which the highest current output was recorded.
Fig. 4 shows a characteristic amperometric response of Pt/GOx-PVA to glucose in the range 0.1–37 mM at −400 mV in buffer solution. For a comparison, also amperometric responses of bare Pt, of Pt/FAD-PVA and of Pt/PVA are shown. It can be observed that the biosensor Pt/GOx-PVA exhibited the highest current at all tested concentrations confirming the excellent electrocatalytic behaviour of the developed system. Bare Pt and Pt/FAD-PVA electrodes allowed in fact detecting only glucose concentrations higher than about 1 mM, producing not reproducible current signals affected by low signal/noise ratio. The PVA membrane alone prevented the direct oxidation of the glucose at the electrode surface, as revealed by the absence of any significant oxidation current in all the explored concentration range.
 | ||
| Fig. 4 Amperometric responses of Pt/GOx-PVA (red curve), Pt/FAD-PVA (black curve), bare Pt (blue curve) and Pt/PVA (green curve) to glucose 0.1–37 mM in stirred and nitrogen saturated PBS (pH 7.0, I = 0.2). A zoom of Pt/GOx-PVA response to glucose 0.1–3.5 mM is reported in the inset. Measurements were performed in PAD mode (600 mV for 0.2 s; −630 mV for 0.2 s; detection potential: −400 mV). | ||
On Pt/GOx-PVA a very low limit of detection (LOD) was observed being equal to 10 μM. It showed also a very rapid time response of about 4 s. This quick response was likely due to the direct contact between the electrode surface and the GOx immobilized on it. Also the role of the PVA membrane was studied by performing experiments on a Pt electrode with only GOx directly adsorbed on it.60 No amperometric response was observed in the adopted experimental conditions confirming also that the PVA film is fundamental in determining the catalytic response.
A typical glucose calibration curve (referred to measurements performed in triplicate on three different electrodes) in the range 0.1–37 mM at a Pt/GOx-PVA biosensor is reported in Fig. 5a. The recorded response showed, as expected, saturation at higher glucose concentration due to the saturation of the enzyme active sites. A linear relationship was observed at low substrate concentrations in the range 0.3–2.0 mM (R2 = 0.9913) with a sensitivity of 9.66 μA mM−1. The repeatability of the current response was investigated at glucose concentrations of 1, 5 and 10 mM (three successive assays) and revealed to be equal to 2.9%, 10% and 4%, respectively. Also a good reproducibility was obtained for responses to glucose 1, 5 and 10 mM recorded on three Pt/GOx-PVA films prepared independently being equal to 4.6%, 8.1% and 4.5%, respectively.
 | ||
| Fig. 5 (a) Calibration curves on Pt/GOx-PVA for response to glucose 0.1–37 mM. The error bars refer to measurements performed in triplicate on three different electrodes. Experimental conditions are as in Fig. 4. (b) Plot of 1/Ivs. 1/C for Pt/GOx-PVA response. I represents the current recorded after each glucose addition and C is the glucose concentration. | ||
A linear relationship between the reciprocal of current response (1/I) and the reciprocal of glucose concentration (1/C) was obtained in the concentration range of 2.0–37 mM (Fig. 5b). According to the Lineweaver–Burk equation, the slope of the linear fit has been used to evaluate K′m obtaining a value of 12.8 mM. K′m value was much smaller than that reported for other mediatorless glucose biosensors based on GOx immobilized in TTF-TCNQ (33 mM),18 in a sol–gel chitosan composite (21 mM),62 on the surface of nano-CaCO3 (21.4 mM)63 and also for GOx in solution (22 mM).63 These results clearly suggest that GOx displays a higher affinity for glucose in the developed system.
Electrode performance was revealed to be very satisfactory in terms of detection limit, linear range, response time and sensitivity, even if compared with nanotechnology-based systems for glucose sensing64 typically employing a more time-consuming scheme and more expensive materials for electrode preparation.
3.4 Selectivity study
Possible interference from coexisting species with glucose in biological fluids (ascorbic acid and uric acid) and from other sugars (fructose and sorbitol) has been studied by analyzing PAD biosensor response to each of the interference molecules. Results are shown in Fig. S7 (see ESI†). As can be seen, fructose, sorbitol and uric acid gave undetectable signals, while ascorbic acid determined only a slight decrease of glucose oxidation current (about 7%).With the aim to further confirm that FAD linked to protein was responsible for the observed electrochemical behaviour on the Pt/GOx-PVA electrode, the amperometric response to interfering substances was analyzed also on the Pt/FAD-PVA electrode in the same experimental conditions. Fructose, uric acid and ascorbic acid, respectively, produced a reduction current 2-, 3- and 2.5-fold higher than the glucose oxidation signal, while the sorbitol oxidation current was comparable to the glucose current response. These results allowed us to confirm that the electroactivity of the Pt/GOx-PVA electrode is due to protein in its intact structure and not to free FAD molecules released from the enzyme. Evidently, also biosensor selectivity and specificity is attributable to enzyme structural integrity.
3.5 Biosensor storage stability
Two methods were used to store the Pt/GOx-PVA electrode. One was to place it in PBS (pH 7.0, I = 0.2) at +4 °C when not in use. Biosensor response stability was evaluated by monitoring its amperometric response to glucose in the concentration range 0.1–37 mM after 3, 5 and 35 days following biosensor preparation. An average response decrease of 20%, 32% and 39% was observed, respectively.Alternatively, the electrode was left dry at room temperature in a closed vial until further experiments and the response stability was evaluated as described above. Surprisingly, in this case, the average biosensor response decrease was only about 13% after 35 days, evidencing a very satisfactory stability under such storage conditions similar to or better than that reported for other glucose biosensors.65–68
3.6 Evaluation of biosensor stability by XPS analysis
Survey XPS spectra in Fig. 6(i) show, for each Pt/GOx-PVA film (a–d) and for PVA film (e), as expected, a surface with intense C1s and O1s signals and without sodium phosphate contamination even after being used as a sensor (c and d spectra). | ||
| Fig. 6 Wide XPS spectra (i), Pt4f detailed spectrum (ii) and valence band VB (iii) for Pt/GOx-PVA: after its preparation (a), after 10 CV cycles in clean PBS (b) and in PBS with glucose 10 mM (c), after amperometric measurements in PAD mode (d). Spectra (e) are referred to Pt/PVA. For details see Experimental. | ||
Detailed spectra in the N1s region showed, in all the Pt/GOx-PVA films, the presence of component peaks characteristic of the GOx standard. In particular, the N1s signal was present also in the spectrum relevant to film used in PAD experiments (d spectrum).
Analysis of detailed spectra of Na1s and P2p regions confirms that only very small contamination was present on the film surface. Moreover, detailed spectra of Pt4f showed the absence of the signal relative to Pt4f on each sample even after using the film for glucose detection (Fig. 6(ii), spectra c–d). Both these data confirm a compact deposition of films on the Pt substrate not modified after either CV or amperometric measurements.
Finally, also the valence band (VB) region for these samples (Fig. 6(iii)) is clearly representative of PVA films.69 All the collected data confirm that, even after being used for glucose detection, the film is still compact and uniformly distributed on the Pt surface.
These spectroscopic data, in agreement with electrochemical results, confirm that the sensor response is not directly related to the Pt substrate only but it is due to a synergistic effect between Pt and the film GOx-PVA.
Conclusions
The present work describes the development of a new mediatorless glucose biosensor obtained by the entrapment of GOx in a PVA matrix on a Pt substrate. GOx structural integrity in PVA was verified by UV-vis as well as enzymatic activity towards glucose oxidation in the adopted experimental conditions. Electrochemical investigations of the system by CV with and without glucose revealed that the glucose oxidative process was supported by the redox conversion of the FAD/FADH2 center of GOx that involved a direct electron transfer with the Pt surface to give hydrogen protons de-adsorption. The comparison of redox properties of GOx- and of FAD-modified electrodes allowed us to verify that the electrochemical behaviour of the developed biosensor was due to FAD linked to the protein, thus confirming the spectroscopic results on enzyme structural integrity.Satisfactory performance of the biosensor was observed in glucose amperometric detection in PAD mode obtaining low detection limit, wide linear range and good sensitivity and selectivity. Moreover, the low value of calculated K′m evidenced a high biological affinity to glucose. Finally, the stability of the biosensor both under storage and operative conditions was verified by electrochemical and XPS characterization, respectively.
References
- A. Amine, H. Mohammadi, I. Bourais and G. Palleschi, Biosens. Bioelectron., 2006, 21, 1405–1423 CrossRef CAS.
- J. Wang, J. Pharmaceut. Biomed., 1999, 19, 47–53 CrossRef CAS.
- A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS.
- W. Zhang and J. Li, Anal. Sci., 2004, 20, 603–609 CAS.
- G. F. Khan, M. Ohwa and W. Wernet, Anal. Chem., 1996, 68, 2939–2945 CrossRef CAS.
- J. Liu, M. N. Paddon-Row and J. J. Gooding, J. Phys. Chem. B, 2004, 108, 8460–8466 CrossRef CAS.
- C. Malitesta, F. Palmisano, L. Torsi and P. G. Zambonin, Anal. Chem., 1990, 62, 2735–2740 CrossRef CAS.
- J. Wang, G. Rivas and M. Chicharro, Electroanal., 1996, 8, 434–437 CAS.
- M. C. Rodriguez and G. A. Rivas, Electroanal., 2001, 13, 1179–1184 CrossRef CAS.
- A. A. Karyakin, E. A. Kotelnikova, L. V. Lukachova and E. E. Karyakina, Anal. Chem., 2002, 74, 1597–1603 CrossRef CAS.
- Q. Chen, P. V. A. Pamidi, J. Wang and W. Kutner, Anal. Chim. Acta, 1995, 306, 201–208 CrossRef CAS.
- E. Kosela, H. Elzanowska and W. Kutner, Anal. Bioanal. Chem., 2002, 373, 724–734 CrossRef CAS.
- F. Patolsky, Y. Weizmann and I. Willner, Angew. Chem. Int. Ed., 2004, 43, 2113–2117 CrossRef CAS.
- U. Anik, S. Timur, M. Cubuku and A. Merkoci, Microchim. Acta, 2007, 160, 269–273.
- M. Viticoli, A. Curulli, A. Cusma, S. Kaciulis, S. Nunziante, L. Pandolfi, F. Valentini and G. Padeletti, Mater. Sci. Eng. C, 2006, 26, 947–951 CrossRef CAS.
- C. Zhao, Y. Meng, C. Shao, L. Wan and K. Jiao, Electroanal., 2008, 20, 520–526 CrossRef CAS.
- F. Palmisano, P. G. Zambonin, D. Centonze and M. Quinto, Anal. Chem., 2002, 74, 5913–5918 CrossRef CAS.
- M. Cano, J. L. Avila, M. Mayen, M. L. Mena, J. Pingarron and R. Rodriguez-Amaro, J. Electroanal. Chem., 2008, 615, 69–74 CrossRef CAS.
- J. Wu and Y. Qu, Anal. Bioanal. Chem., 2006, 385, 1330–1335 CrossRef CAS.
- C. G. J. Koopal, B. de Ruitter and R. J. M. Notle, J. Chem. Soc., Chem. Commun., 1991, 1691–1692 RSC.
- A. Guiseppi-Elie, C. Lei and R. H. Baughman, Nanotechnology, 2002, 13, 559–564 CrossRef.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS.
- J. Zhang, M. Feng and H. Tachikawa, Biosens. Bioelectron., 2007, 22, 3036–3041 CrossRef CAS.
- J. Wu, Y. Zoua, N. Gaoa, J. Jiang, G. Shen and R. Yu, Talanta, 2005, 68, 12–18 CrossRef CAS.
- M. Zhao, X. Wu and C. Cai, J. Phys. Chem. C, 2009, 113, 4987–4996 CrossRef CAS.
- D. Shan, J. Zhang, H. Xue, S. Ding and S. Cosnier, Biosens. Bioelectron., 2010, 25, 1427–1433 CrossRef CAS.
- Y. Degani and A. Heller, J. Phys. Chem., 1987, 91, 1285–1289 CrossRef CAS.
- F.-L. Wong and A. Adbdul-Aziz, J. Chem. Technol. Biot., 2008, 83, 41–46 Search PubMed.
- L. Doretti, D. Ferrara, P. Gattolin, S. Lora, F. Schiavon and F. M. Veronese, Talanta, 1998, 45, 891–898 CrossRef CAS.
- D. Ivnitski, K. Artyushkova, R. A. Rincon, P. Atanassov, H. R. Luckarift and G. R. Johnson, Small, 2008, 4, 357–364 CrossRef CAS.
- G. Zoldak, A. Zubrik, A. Musatov, M. Stupak and E. Sedlak, J. Biol. Chem., 2004, 279, 47601–47609 CrossRef.
- H. Y. Liu and N. F. Hu, Electroanal., 2007, 19, 884–892 CrossRef CAS.
- H. Yang, P. M. Johnson, K. C. Ko, M. Kamio, M. W. Germann, C. D. Derby and P. C. Tai, J. Exp. Biol., 2005, 208, 3609–3622 CrossRef CAS.
- A. J. Bard, L. R. Faulker, Electrochemical Methods, Wiley, New York, 2nd edn, 2001 Search PubMed.
- P. N. Jr. Ross, J. Electrochem. Soc., 1979, 126, 67–77.
- S. Park, H. Boo and T. D. Chung, Anal. Chim. Acta, 2006, 556, 46–57 CrossRef.
- S. Ernst and J. Heitbaum, J. Electroanal. Chem., 1979, 100, 173–183 CrossRef CAS.
- T.-H. Yang, C.-L. Huang, J.-H. Ke and J.-M. Zen, Electrochem. Commun., 2008, 10, 1094–1097 CrossRef CAS.
- C. Cai and J. Chen, Anal. Biochem., 2004, 332, 75–83 CrossRef CAS.
- H. Durliat, M. B. Barrau and M. Comtat, Bioelectrochem. Bioenerg., 1988, 19, 413–423 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1995, 382, 111–127 CrossRef.
- F. R. Duke, R. N. Kust and L. A. King, J. Electrochem. Soc., 1969, 116, 32–34 CrossRef CAS.
- F. Patolsky, G. Tao, E. Katz and I. Willner, J. Electroanal. Chem., 1998, 454, 9–13 CrossRef CAS.
- S. Demin and E. A. H. Hall, Bioelectrochemistry, 2009, 76, 19–27 CrossRef CAS.
- Z. Wen, B. Ye and X. Zhou, Electroanalysis, 1997, 9, 641–644 CAS.
- H. J. Lowe and W. M. Clark, J. Biol. Chem., 1956, 221, 983–992 CAS.
- C. N. Durfor, B. A. Yenser and M. L. Bowers, J. Electroanal. Chem., 1988, 244, 287–300 CrossRef CAS.
- M. F. J. M. Verhagen and W. R. Hagen, J. Electroanal. Chem., 1992, 334, 339–350 CrossRef CAS.
- A. Niemz, J. Imbriglio and V. M. Rotello, J. Am. Chem. Soc., 1997, 119, 887–892 CrossRef CAS.
- S. Liu and H. Ju, Biosens. Bioelectron., 2003, 19, 177–183 CrossRef CAS.
- W. Zhang, Y. Huang, H. Dai, X. Wang, C. Fan and G. Li, Anal. Biochem., 2004, 329, 85–90 CrossRef CAS.
- K. M. Manesh, H. T. Kim, P. Santhosh, A. I. Gopalan and K.-P. Lee, Biosens. Bioelectron., 2008, 23, 771–779 CrossRef CAS.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 CrossRef CAS.
- P. Andrieux and J. M. Saveant, J. Electroanal. Chem., 1978, 93, 163–168 CrossRef CAS.
- M. Yoshimoto, M. Sato, S. Wang, K. Fukunaga and K. Nakao, Biochem. Eng. J., 2006, 30, 158–163 CrossRef CAS.
- S. Thibault, H. Aubriet, C. Arnoult and D. Ruch, Microchim. Acta, 2008, 163, 211–217 CrossRef CAS.
- S. B. Bankar, M. V. Bule, R. S. Singhal and L. Ananthanarayan, Biotechnol. Adv., 2009, 27, 489–501 CrossRef CAS.
- A. Martins, V. Ferreira, A. Queirós, I. Aroso, F. Silva and J. Feliu, Electrochem. Commun., 2003, 5, 741–746 CrossRef CAS.
- G. E. De Benedetto, C. Malitesta and C. G. Zambonin, J. Chem. Soc., Faraday Trans., 1994, 90, 1495–1499 RSC.
- X. Chen, J. B. Jia and S. Dong, Electroanalysis, 2003, 15, 608–612 CrossRef CAS.
- D. Shan, M. Zhu, H. Xue and S. Cosnier, Biosens. Bioelectron., 2007, 22, 1612–1617 CrossRef CAS.
- B. Wang, B. Li, Q. Deng and S. Dong, Anal. Chem., 1998, 70, 3170–3174 CrossRef CAS.
- J. C. Claussen, A. D. Franklin, A. Haque, D. M. Porterfield and T. S. Fisher, ACS Nano, 2009, 3, 37–44 CrossRef CAS.
- Y. Y. Horng, Y. K. Hsu, A. Ganguly, C. C. Chen, L. C. Chen and K. H. Chen, Electrochem. Commun., 2009, 11, 850–853 CrossRef CAS.
- P. Benvenuto, A. K. M. Kafi and A. Chen, J. Electroanal. Chem., 2009, 627, 76–81 CrossRef CAS.
- Y. Zhang, G. Guo, F. Zhao, Z. Mo, F. Xiao and B. Zeng, Electroanalysis, 2010, 22, 223–228 CrossRef CAS.
- X. Chu, B. Wu, C. Xiao, X. Zhang and J. Chen, Electrochim. Acta, 2010, 55, 2848–2852 CrossRef CAS.
- D. J. Asunskis and L. Hanley, Surf. Sci., 2007, 601, 4648–4656 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures S1-S7. See DOI: 10.1039/c0an00194e |
| This journal is © The Royal Society of Chemistry 2011 |