Protease sensing with nanoparticle based platforms
Katharina
Welser
ab,
Rosemary
Adsley
ab,
Bernadette M.
Moore
b,
Weng C.
Chan
*a and
Jonathan W.
Aylott
*b
aSchool of Pharmacy, Centre for Biomolecular Sciences, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: weng.chan@nottingham.ac.uk
bSchool of Pharmacy, Boots Science Building, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: jon.aylott@nottingham.ac.uk
First published on 28th September 2010
Abstract
Nanoparticulate systems in various unique configurations are highly effective at detecting protease activity both in vivo and in vitro. In this article, we have summarised the conventional modern methods for monitoring protease activity, and critically appraised recent advances in protease-responsive nanosensors.
 Katharina Welser | Katharina Welser obtained her MSc in Chemistry from the Technical University Graz for her work centered on the design of photoswitchable immobilization matrices for biochips. She then moved to the University of Nottingham, where she was awarded a PhD in Pharmacy for the design of smart optical imaging agents for protease detection under the guidance of Dr Jonathan Aylott and Dr Weng Chan. After her PhD, she worked as a postdoctoral researcher on drug delivery systems at the University of Nottingham, before taking up her current research associate position at University College London, where she focuses on siRNA delivery and targeting strategies. |
 Rosemary Adsley | Rosemary Adsley received her Pharmacy degree from the University of Bath in 2007 and carried out pre-registration training with Pfizer and Alliance Pharmacy, qualifying in 2008. She is currently undertaking doctoral studies at the University of Nottingham as a member of the AstraZeneca-EPSRC Targeted Therapeutics Doctoral Training Centre. Her research is focussed on using novel imaging techniques to investigate the cellular uptake of nanoparticles. |
 Weng C. Chan | Dr Weng Chan received his PhD degree from the University of Nottingham in 1988, followed by postdoctoral training (1988–1992) under the guidance of Professors Barrie Bycroft and Gordon Roberts. He is currently Associate Professor and Reader in Chemical Biology. He has (co)authored over 80 research and review articles, patents, book chapters and conference proceedings. His research is focussed on the design and synthesis of unique chemical constructs, including responsive nanomaterials, for interrogating and modulating biomolecular processes, especially those involved in bacterial pathogenesis, carcinoma and neurodegenerative diseases. |
 Jonathan W. Aylott | Jonathan Aylott is a Lecturer in Bioanalytical Science in the School of Pharmacy at the University of Nottingham. He received his PhD in Analytical Chemistry from the University of East Anglia for the development of optical biosensors using sol–gel immobilised metalloproteins. He then undertook postdoctoral training at the University of Michigan working on miniaturized optochemical sensors with Professor Raoul Kopelman. Upon returning to the UK he took a lectureship at the University of Hull, before moving to his current role. His research is focused on the development and application of optical nanosensors for the measurement of biological systems. |
1. Introduction
Over the last decade nanoparticles have proven to be extremely convenient and invaluable tools for biological and biomedical applications. In addition to serving as excellent drug delivery vehicles and bio-imaging agents,1–4nanoparticles have been used to create biosensing devices of extremely high sensitivity. Among the most important sensing targets are proteases, as they are not only extensively involved in biological processes such as hormone activation and apoptosis, but are also implicated in many diseases including AIDS and cancer.5–9 A wide variety of nanoparticles have been developed in recent years that have the potential to be used as biosensing devices viaconjugation to relevant proteins and peptides, as well as fluorophores and chromophores for imaging purposes. Most have been shown to be non-toxic in vitro, suggesting that further in vivo applications may be possible. In this perspective, we discuss and compare recent advances in the emerging field of nanoparticulate-based systems for the detection of proteases in vitro and in vivo.2. The role of proteases in disease states
Proteases are enzymes that selectively cleave peptide bonds in proteins and polypeptidesviahydrolysis.10–13 At least 500–600 proteases (∼2% of the genome) have been identified by bioinformatic analysis of the mouse and human genome.13 Proteases play a crucial role in a wide variety of processes within the body including blood coagulation, hormone activation and maturation, protein digestion, apoptosis, fertilization, growth differentiation and cell signalling.10,12,13 Apart from playing an important role in the post-translational modification of proteins,14–16 they also help to regulate protein turnover by breaking down proteins into their constituent amino acids and peptide fragments.12Furthermore, proteases are key participants in a number of disease states including viral infections such as hepatitis and AIDS,17,18 cardiovascular disease,19 cancer,20 Alzheimer's disease21–23 and inflammatory diseases.24–28 Several drugs act by modulation of protease activity, e.g.angiotensin converting enzyme inhibitors such as ramipril, used to treat hypertension, and protease inhibitor drugs used in the treatment of HIV infections such as ritonavir.12
In the field of oncology, there is a great deal of interest in examining the possibility of inhibiting proteases, including matrix metalloproteinases (MMPs) and cathepsins, which play a role in metastasis.20,24,29,30 A common feature of these protease modifying drugs is their specificity for the targeted protease without affecting other proteases in the body.12,13
Investigation into the detection and real time monitoring of proteases in vitro and in vivo is of great interest as proteases can be used as markers of certain diseases.13 One of the best examples is the detection of the serine protease kallikrein 3, also known as PSA (prostate specific antigen), which is an important diagnostic marker for prostate cancer.13,31
Research on apoptosis revealed the key role of caspases in programmed cell death. Targeting these caspases is especially interesting as inappropriate caspase activity or overexpression of caspase inhibitory molecules has been found in various types of cancer cells.32 The cysteine protease cathepsin B, found upregulated in breast cancer, has been associated with tumor progression,33,34 while cathepsin D has been reported as a potential biomarker for breast cancer.35 Another important protease in tumor progression is the urokinase plasminogen activator, which plays a role in proteolytic activation of plasminogen to plasmin that further catalyzes downstream activation of several matrix metalloproteases.36
Hyperactivation of calpains, a family of intracellular cysteine proteases, is implicated in various pathologies associated with altered calcium homeostases, including myocardial injury, brain ischemia, Duchenne muscular dystrophy and Alzheimer's and Huntington's neurodegenerative disorders.37
A great deal of research has thus been devoted to develop technologies that allow the direct visualization of protease activity in cells and small animals in real time. These technologies involve both the design of smart optical imaging reagents and the development of optical instrumentation that allow the sensitive and rapid detection of enzyme activity within cells and whole organism.38 In recent years, conventional molecular tags such as fluorophores have increasingly being replaced by nanoparticles such as quantum dots, as they offer several advantages including superior optical properties, substantially greater chemical stability and stability against photobleaching.39–43 One major merit of using some nanomaterials is that their optical properties can systematically be varied via modification of particle size or dimension, leading to an array of new applications.39,44,45 In the following, the most important conventional and nanoparticle based technologies are described which provide a direct readout of enzyme activity.
3. Conventional protease assays
Small molecule substrate reporters of protease activity
Fluorogenic substrates have been widely used in order to detect enzymatic activity. Virtually all function using a mechanism in which the fluorescence of the reporter molecule increases and/or changes after proteolytic cleavage of the peptidic scaffold. The use of short synthetic substrates to detect protease activity has several advantages over using whole proteins as substrate. For instance, each peptide bond in a protein acts as a potential substrate for different proteases. As proteolysis proceeds, these substrates and their accessibility change as the protein is broken down to smaller peptides, resulting in a different susceptibility of the peptide bonds to be cleaved. Factors such as pH influence the charge properties of the protein substrate as well as the enzyme, and may therefore critically influence substrate binding. In contrast, synthetic peptide substrates are designed so that only a specific bond is cleaved and the progress of proteolysis can be monitored continuously usually by spectroscopic means.46 Three major types of activatable fluorescent probes have been developed to report protease activity in living systems (Fig. 1):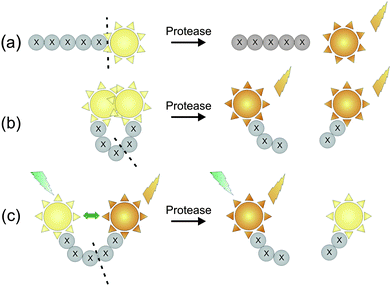 | ||
| Fig. 1 Different designs of fluorescent substrate reporters for the imaging of protease activity. (a) Reporter substrate based on a quenched peptide-bound fluorophore. (b) Reporter substrate in which the fluorophore is quenched by close proximity to a quencher molecule. (c) Reporter substrate based on fluorescence resonance energy transfer (FRET) (adapted from ref. 49). | ||
Protein-based fluorescent and bioluminescent probes
The green fluorescent protein (GFP) originally isolated from the Pacific jellyfish Aequorea victoria has revolutionised many aspects of cell biology and has become a key component of many protein-based probes designed to image molecular and cellular events.49,59,60 For example, the use of GFP has enabled the real time monitoring of in vivo proteolytic events mediated by a variety of proteases including Factor Xa,61 trypsin,62caspase-1 and caspase-3.63–65 Using rational/or random mutation techniques, many GFP variants have been produced that differ in protein and chromophore structure and hence in their absorption and emission profiles. Examples of such fluorescent protein variants include a more enhanced and stable GFP, as well as blue, cyan, yellow and red fluorescent proteins (BFP, CFP, YFP and RFP).57Fluorescent proteins have found extensive use in FRET-based probes, as FRET can occur between their spectral variants. The most commonly used FRET pair consists of the cyan fluorescent protein (CFP) and the yellow fluorescent protein (YFP),11,49,56,60,66,67 although recently red fluorescent proteins (RFPs) have proven their utility as FRET-based reagents.49 A general example for a FRET-based reporter used for the detection of intracellular protease activity is shown in Fig. 2. FRET occurs between a donor CFP and an acceptor YFP which are linked by a spacer containing a protease cleavable substrate. The presence of the protease results in cleavage of the linker, and the consequent spatial separation of the FPs and the loss of measurable energy transfer. Using this probe design it was possible to visualize the execution of apoptosis in a single cell by monitoring the caspase-3 mediated cleavage of a linker containing a DEVD sequence.67 More recently, Stockholm et al. reported a calpain sensor composed of eCFP (enhanced cyan fluorescent protein) and eYFP (enhanced yellow fluorescent protein), linked together by a calpain cleavage site. Activation of calpain by Ca2+ resulted in cleavage of the substrate, inducing separation of the two fluorescent reporters and disappearance of FRET. This probe was used to efficiently and quantitatively monitor calpain activation in living mouse tissues at a cellular level.37

However, the fluorescence of both CFP and YFP is weak compared with the wild-type GFP.49 In addition, GFP-based constructs can suffer from limited sensitivity, and due to their large size, can limit spatial resolution.68 It has also been reported that GFP can undergo colour changes upon irradiation due to photochemical changes that are independent of FRET.49,68 A general limitation of FRET based probes is the requirement for external illumination to initiate fluorescence transfer causing background noise or photobleaching.69 To try and avoid some of these problems, protein probes based on bioluminescence energy transfer (BRET) have been developed.
Bioluminescence is a natural phenomenon where energy is released by a chemical reaction in the form of light emission. The key step is the oxidation of a substrate molecule (luciferin) by enzyme (luciferase) generating energy rich peroxide intermediates. Spontaneous decomposition of these intermediates forms product molecules in the excited state which relax back to the ground state by emitting light.69 In BRET, the fluorescent donor is replaced by a bioluminescent luciferase. This bioluminescent protein produces an initial photon emission, which is then transferred via a nonradiative process to an acceptor, for example a fluorescent protein that absorbs the donor energy and emits light at a longer wavelength.70 Widely utilized luciferases are derived from the firefly (Photinus pyralis) and sea pansy (Renilla reniformis). Even though fluorescent signals are brighter than bioluminescent ones, allowing a better resolution in cells, bioluminescent reporters have the advantage of high sensitivity, combined with minimal autoluminescence of cells and tissues.69,71 For instance, a direct comparison between a representative FRET system (CFP donor and YFP acceptor) and a BRET system (Renilla luciferase donor, GFP acceptor and coelenterazine 400a substrate), which were used for the detection of thrombin, showed that the BRET probe was 50 times more sensitive than the FRET system.56
One disadvantage of the use of proteins in RET systems is that they have small Stokes shifts, which results in poor spectral separation of the acceptor emission from the donor emission. This problem might be overcome by the use of a new class of fluorophores based on semiconductor materials, quantum dots (QDs), which possess a large Stokes shift.70
Another bioluminescent reporter system, which enabled imaging of caspase-3 activity in cells undergoing apoptosis, was reported by Laxman et al.71 The probe (Fig. 3) consists of a luciferase fusion protein flanked by two estrogen receptor (ER) regulatory domains which are linked through a caspase-3 cleavable linker. In the absence of caspase-3, luciferase activity is silenced due to the steric effects mediated by the binding of heat-shock proteins to the ER domains. The luciferase activity is restored by the cleavage of the ER silencing domain by caspase-3, resulting in a 10-fold increase in luminescence. This technology was used in order to monitor caspase-3 activity in reporter-expressing tumors implanted into mice in real-time.
 | ||
| Fig. 3 A bioluminescent reporter for in vivo imaging of caspase-3 activity (adapted from ref. 71). HSP = heat shock protein and ER = estrogen receptor regulatory domains. | ||
More recently, a system was introduced which incorporated the luciferin into the assay design in a similar manner to small molecule substrate assays. The assay uses an aminoluciferin, which is modified by a Z-DEVD group and therefore becomes unavailable to the luminescent reaction.72 However, cleavage of the tetrapeptide by the target protease (caspase-3) ‘activates’ the luciferin, thus allowing its further oxidation by luciferase which results in a detectable luminescent signal. When compared under similar conditions to a fluorescent assay, the bioluminescent system was nearly 100-fold more sensitive (Fig. 4b). The increased sensitivity is due to the greatly reduced background.
 | ||
| Fig. 4 Comparison between bioluminescent and fluorescent measurement of caspase-3/7 activity. (a) Assay scheme of caspase cleavage on Z-DEVD aminoluciferin. (b) Assays were performed on Jurkat cells treated with anti-FAS and serially diluted into 96-well plates. Measurements were made 1 hour after reagent addition. Results were plotted as signal to noise ratios. (Reprinted with permission from O'Brien.72) | ||
Polymer-based near infrared fluorescence probes
A number of polymer-based near-infrared fluorescence (NIRF) probes have been introduced by the Weissleder group that allow the noninvasive visualization of enzyme activity in whole organism.24,73 The probes consist of a high molecular weight carrier, a cleavable peptide spacer, and a (NIR) fluorophore (Fig. 5). NIR probes are typically designed to be maximally intramolecularly quenched in the intact state and to be brightly fluorescent when proteolytically released from the polymer. The probes are ideal for in vivo imaging due to the high penetration of photons emitted by the NIR fluorochrome, the low autofluorescence of non-target tissue in the NIR spectrum (650–900 nm), their prolonged plasma-half lives, enhanced stability and reduced nonspecific binding.74 Most NIRF probes are based on indocyanine green, a tricarbocyanine dye approved by the FDA for clinical ophthalmic retinal angiography, cardial function and liver function testing.74 A typically used high molecular weight carrier in NIR probes is polyethylene glycol (PEG) protected graft co-polymer (PGC), originally developed for drug targeting. Protease-sensing PGC-based NIR probes have been designed to monitor proteases such as cathepsin,75 metalloproteases,76,77 and coagulation enzymes78in vivo. Although these PGC based probes have been proven to be very useful for in vivo imaging, several disadvantages limit their application.73 For example, even though the PEG chain affords the probe increased stability, thus allowing longer circulation time, the PEG chain might also disturb the interaction between the target enzyme and the peptide substrate.74 Such an interference could result in a significant decrease in the sensitivity of the probe. Additionally, the NIR fluorophores might not be cleaved as efficiently by the target protease as they are tightly conjugated to the PGC backbone.74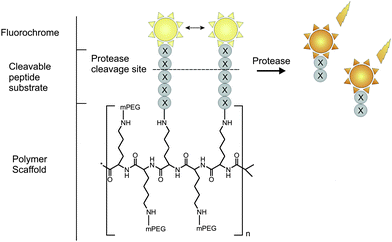 | ||
| Fig. 5 Peptide-based fluorescent reporter for protease sensing with a NIRF fluorochrome. | ||
Antibody-based protease probes
Enzyme-linked immunosorbent assays (ELISA) have been reported for the detection of HIV-1 and several MMP family members.79–81 The general principle of a conventional solid-phase indirect sandwich ELISA is given in Fig. 6a. For this assay,79,82 three antibodies (Ab) are needed: (1) one to immobilize the protease, typically a pAb; (2) one to detect the protease, typically a mAb; and (3) a secondary antibody to detect the mAb, which is additionally linked to an enzyme. Detection is achieved colorimetrically by conversion of an exogenous substrate or by the use of a fluorescently labelled secondary Ab, and hence does not provide functional information. A modified and improved version of this assay was published by Fields and co-workers for the detection of MMP-1, MMP-3, MMP-13 and MMP-14.79 The assay consisted of two principal components: a capture antibody that traps the MMP without perturbing the active site, and a fluorescence resonance energy transfer substrate for measuring proteolysis at low enzyme concentrations (Fig. 6b). It was shown that the assay had several advantages over prior methods for analyzing MMP activity, including (a) enzyme selectivity, (b) high sensitivity, (c) responsive to enzymatic activity and (d) amenable to high-throughput analysis. | ||
| Fig. 6 Schematic illustration of (a) conventional indirect sandwich ELISA and (b) solid-phase MMP activity assay. In both assays, the MMP is captured by an antibody on the solid phase. A further detection antibody and an enzyme-labelled antibody is required for the indirect sandwich ELISA. The sandwich ELISA can also be performed directly by using an enzyme labelled detection antibody. For the solid-phase MMP activity assay, the fluorogenic substrate serves as the detection system. Mca, (7-methoxycoumarin-4-yl)acetyl and Dnp, (2,4-dinitrophenyl).79,82 | ||
Activity-based probes
The main disadvantage of the substrate-based imaging methods described so far is the potential lack of specificity toward enzyme targets. In fact, a peptide has the potential to serve as substrate for more than one class of proteolytic enzymes. An alternative approach for studying the role of proteases in pathological processes is the use of activity-based probes (ABPs).29,49 These are small molecules that covalently bind to an enzyme-target using chemical interactions that are specific for the target enzyme. ABPs generally contain three main functional groups: a reactive group or warhead, a linker region and a visualization tag (Fig. 7). Probably the most crucial component is the warhead, which covalently reacts with amino acid residues of the target enzymes. To target an ABP to a specific subset of enzymes, a linker region is used, which mimics a true substrate, while the tag group allows the detection or isolation of the modified enzyme.83 One of the major limitations of the use of fluorescent ABPs in cell-based imaging applications is that they produce a fluorescent signal both when they are bound to the target enzyme and when they are free in solution. To address this problem quenched activity-based probes (qABPs) were designed, in which a quencher molecule was installed in close proximity to the fluorophore to prevent fluorescence emission. However, fluorescence is restored when the qABP covalently binds to the target enzyme, due to the loss of the quencher as part of the leaving group resulting from the nucleophilic substitution reaction with the active side residue. These probes, which were successfully used for the in vivodetection of cysteine protease and cathepsin, only emit fluorescence when bound to the target enzyme.22,23 | ||
| Fig. 7 General structure of an activity based probe (ABP). | ||
4. Nanoparticle-based protease sensing strategies
A wide variety of nanoparticle based systems have been developed with varying degrees of success and sensitivity. Detection can be performed using fluorometric methods, surface-enhanced Raman techniques, relaxation measurements, absorbance or visual detection.84 Typically, nanoparticle enzyme sensors consist of a biological substrate specific to the enzyme of interest, immobilised onto the nanoparticle surface. The active enzyme induces a change in the substrate composition or conformation, imparting a change in the environment of the nanoparticle, which results in a detectable signal change. For example, systems that incorporate fluorogenic substrates generally act by detecting a change in the fluorescence of a reporter molecule following cleavage of the peptide substrate by the protease.85–87 These nanoparticulate systems are useful tools to monitor an enzyme system in real time and may provide a useful high-throughput screening tool for enzyme activity and drug discovery.84Some of these systems are outlined below and their benefits and disadvantages discussed. Each system uses different materials to produce a nanoscale device that is capable of sensing and monitoring protease activity both in vitro, and sometimes in vivo, and often with an extremely high degree of sensitivity.
4.1 Protease sensing with inorganic nanoparticles
 | ||
| Fig. 8 Colour changes observed upon the aggregation and dispersion of gold nanoparticles in solution (adapted from ref. 15). | ||
A simple and highly sensitive method for the detection of proteolytic enzymes based on the protease-triggered dispersion of AuNP assemblies was reported by Laromaine et al. (Fig. 9).90 In this example, Fmoc-protected peptides (substrates for thermolysin) were used which contained a cysteine amino acid that was tethered to AuNPs. Upon proteolysis of the peptide–AuNP conjugate, disassembly occurs and the solution colour changes from blue to red.
 | ||
| Fig. 9 (a) Schematic representation of Fmoc-peptide/gold nanoparticle protease sensing system. (b) TEM image of AuNPs after functionalization with peptide (left) and following addition of thermolysin (right). (Reprinted with permission from Laromaine et al.90) | ||
Biocompatible AuNPs are also popular for their NIRF quenching properties, which make them especially attractive for in vivo imaging.91 It is known that fluorophores in close proximity to AuNPs experience strong electronic interactions with the surface, which causes a transfer of excited electrons to the metal nanoparticles. This donation of electrons leads to a quenching in the fluorescence, providing an almost perfectly quenched state.92 Although the precise mechanism of quenching is not clear, the quenching efficiency of AuNPs is likely to be dependent on their size and shape, the distance from the fluorophore, spectral overlap, and dipole orientation.93 Based on this quenching phenomenon, several energy transfer-based sensing systems for the detection of proteases were designed, where proteolytic cleavage of the peptide substrate results in a recovery of the previously AuNP-quenched fluorescence of the fluorophore.85 For example, a simple and robust NIRF-quenched AuNP imaging probe for use in protease inhibitor screening and protease activity determination was published by Lee and co-workers.85 The group designed AuNPs, which were stabilized by Cy5.5 labelled substrates for MMP-2. While the stabilized probe showed strong quenching properties with minimal background signals, the activated probe displayed a strong NIRF signal due to the spatial separation of the AuNP and the fluorophore (Fig. 10a–d). The enzyme selectivity of this AuNP probe was evaluated in vitro and clearly showed that only MMPs were able to recover the potential fluorescence signals. In addition, the authors demonstrated the feasibility of using AuNP probes in the detection and visualization of cancerous cells by targeting MMP-2in vivo in MMP-2 positive bearing mice (Fig. 10b). The described platform can be adopted to any target protease by replacing the specific peptide substrate between the AuNP and the fluorophore.
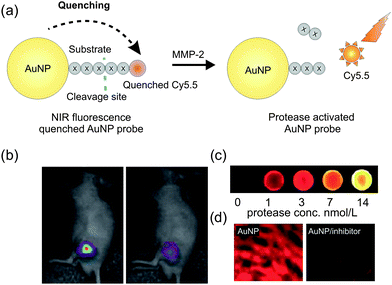 | ||
| Fig. 10 (a) Scheme of a NIRF-quenched AuNP imaging probe. (b) In vivo near-infrared optical imaging of MMPs-positive SCC7 xenografts after intratumoral injection of AuNP probes without (left) or with MMPs inhibitor (right). (c) Fluorescence image from wells containing the AuNP probe in the presence of various concentration of MMP-2. (d) Fluorescence microscopy of SCC7-tumors with AuNP probe that were untreated (left) or treated (right) with inhibitor. (Reprinted with permission from Lee et al.85) | ||
AuNPs possess an extremely high extinction coefficient and broad absorption spectrum.94 The use of AuNPs in energy transfer based systems has the advantage that the fluorescence can be quenched over a wide range of wavelengths.95 Moreover, quenching by AuNPs has often found to occur over much longer distances than those with molecular quenchers and the Förster mechanism.95,96 For instance, QDs and fluorescent dyes have been found to be quenched at distances as large as 16 nm and 21 nm.97,98 This quenching property of AuNPs allows them to be employed as effective proximal quenchers in optical detection. Besides thermolysin90 and MMPs,85 AuNPs have also been used to investigate the activity of thrombin99 and lethal factor from Bacillus anthracis.15
QDs have several obvious advantages over conventional fluorophores.57,103 They are characterized by high quantum yields, prolonged luminescent lifetimes and high molar extinction coefficients that are 10–100 times those of organic dyes.100 Due to their broad absorption spectra, narrow emission profile and large Stoke shifts, a single light source can be used to simultaneously excite multiple species of QDs, thus allowing multiplex sensing. Also, the emission of QDs can be tuned by altering the particle size.44,45,104 Unlike conventional dyes, QDs are extremely resistant to photobleaching, which makes them useful for continuous monitoring of biological phenomena. Additional information on the optical properties of QDs that are relevant for bioimaging can be found from recent review articles.40–43,105–107
QDs have been used for protease sensing using FRET and bioluminescence108 and have been demonstrated in systems coupled to AuNPs.109,110 They have been used to investigate a number of proteases including collagenase,109,111caspase-1,111 thrombin,102 chymotrypsin111 and MMPs.112
Chang et al.109 developed QD probes linked to AuNPs via a peptide sequence that produced an increased luminescence signal in the presence of the protease collagenase.109 Since AuNPs quench fluorescence, a 71% reduction in luminescence was initially observed when QDs were conjugated to AuNPs. Release of the QD by collagenase mediated peptide hydrolysis restored luminescence of the QD.109 The ability to change the peptide linker means that the system can be modified and used to investigate the activity of many different proteases.
Using a similar approach, Medintz and co-workers designed QD probes for different proteases including caspase-1, thrombin, collagenase and chymotrypsin (Fig. 11a).111 Instead of AuNPs, an organic fluorophore or quencher was used as the FRET acceptor. The advantage of this design is that the FRET acceptor is relatively small compared to the gold nanoparticle, which is hypothesised to increase the substrate accessibility to the protease.
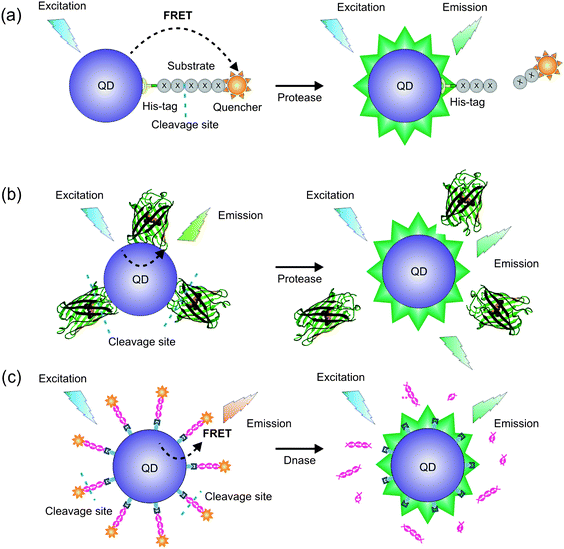 | ||
| Fig. 11 FRET-based QD bioprobes designed to give FRET changes on (a) proteolytic cleavage of the substrate leading to recovery of QD fluorescence;111 (b) cleavage of a GFP variant with an inserted sequence recognized by a protease (e.g. trypsin) to release GFP from the QD surface;113 (c) digestion of dsDNA (labelled with fluorescent dUTP) bound to a QD.113 | ||
Choi et al.102 used thrombin binding aptamer (TBA) capped QDs to detect thrombin activity. Aptamers are single stranded oligonucleotides and are useful since they have a high binding specificity for their target molecule.114,115 The photoluminescence of the QD decreased as thrombin concentration increased, allowing a minimum level of thrombin detection of 1 nM.
Multiplexed sensing offers huge potential in the area of intracellular signalling analysis, such as signalling cascades. To this aim, QD-mounted biosensors were recently described that were capable of operating simultaneously and independently of each other in the same sample solution, allowing the detection of the action of a protease (trypsin) in the presence of deoxyribonuclease (Fig. 11b and c).113 The FRET probes were constructed by immobilizing different fluorescent acceptors such as a mutant of GFP or dsDNA labelled with fluorescent 2′-deoxyuridine-5′-triphosphate (dUTP) onto quantum dots, which act as the corresponding FRET donors. Although it was successfully shown that the sensors could operate simultaneously when excited at a single wavelength, several issues have to be addressed to make these multiplexing biosensors useful for in vitro and in vivo application. It was found that the properties of the bound fluorescent proteins were sometimes be affected by QDs, resulting in function deletion. In addition, the QD excitation profile could be influenced by the attached proteins.
The use of existing quantum dots for in vivo applications is challenging as they require external illumination that generates strong background autofluorescence. Moreover, because of the absorption and scattering of optical photons in tissues, little light is available for QD excitation at non-superficial locations.116 These limitations can be overcome by employing bioluminescence as the source of the resonance energy transfer (BRET) to the QDs. Such ‘self-illuminating quantum dots’ for the sensitive detection of proteases were reported by Yao and colleagues (Fig. 12).108 Using genetic engineering, a MMP-2 specific peptide containing a histidine tag was fused to the C terminus of the BRET donor Luc, which is a mutant of Renillaluciferase. The coordination of nickel(II) ions to the histidine tag led to the association of the engineered protein with the QDs, which further resulted in BRET in the presence of the Luc8 substrate coelenterazine. Treatment of the engineered protein with MMP-2 resulted in proteolytic cleavage of the peptide, preventing the association of the BRET donor with the QD, thus eliminating BRET. Compared to FRET-based QD probes, BRET-based QD biosensors have the advantage that the spectral separation between the BRET donor and acceptor is large. Thus, both emissions can easily be detected and can be used for ratiometric imaging.

However, there are some concerns regarding the use of QDs in biological applications. Questions have been raised regarding the cytotoxicity of inorganic QDs containing Cd, Sn, Hg and Pb. These chemicals can be harmful depending on the dosage, complexation and accumulation in the liver and nervous system and this may limit the suitability of the QDs for in vivo testing.1,100 Thus, it is important to consider the potential cytotoxicity of the core materials when developing nanoscale platforms for protease detection in vivo. New probes such as those not containing Cd and the use of bioluminescence may improve their biocompatibility and enable their use in a clinical setting in future.100
SERS and SERRS approaches have advantages over fluorescence methods. In contrast to fluorescence spectra, which are broad and not unique to the analyte of interest, SERS/SERRS spectra contain sharp vibrational bands, giving fingerprint spectra which are molecularly specific. This means that SERS/SERRS can readily be used to detect multiple different events within a system provided the output signals can, once again, be easily separated.115,121
Typically, surface-enhanced Raman sensors consist of a metallic nanoparticle and enzyme substrates, which consist of three components: (a) an enzyme recognition site to provide specificity, (b) a Raman-active dye and (c) an enzyme-cleavable linker joining the two components. Turnover of the substrate by the enzyme leads to the release of the Raman active species and can either lead to the generation or the disappearance of SERS/SERRS signals which are proportional to the enzyme activity.122–124 Based on this principle, several hydrolytic enzymes including lipases, esterases and proteases have been successfully detected.123
It was demonstrated by Ingram et al. that SERRS can be used to detect the activity of different proteases including chymotrypsin, trypsin and pepsin using silver nanoparticles.14SERRS produced a vibrational spectrum which was characterised by multiple sharp peaks which were equivalent to a ‘fingerprint’ of the Raman dye. Initially, masked protease substrates were used that were transparent to SERRS detection. However, hydrolysis of the substrate led to the release of the Raman active dye and the generation of strong SERRS signals that were proportional to enzyme activity.14
In a further example, Liu and co-workers used SERS for the in vitrodetection of prostate specific antigen (PSA).124 The probe consisted of a nanocrescent particle, to which PSA cleavable substrates with a terminal Raman tag molecule were attached. Cleavage of the substrate by PSA resulted in the spatial separation of the Raman tag molecule from the nanocrescent particles, followed by the disappearance of the Raman tag signal in the SERS spectrum. Compared to other cancer biomarker detection assays, this approach allowed the detection of nanomolar concentrations of proteolytically active PSA in femtolitre volumes, which is crucial especially for cancer screening at a single cancer cell level.124
Two different thrombin sensing systems based on surface enhanced Raman scattering have been developed by the groups of Wang115 and Cho.114 Both sensors use a thrombin binding aptamer (TBA) in combination with a Raman reporter as recognition element for α-thrombin.
Based on the fact that one α-thrombin can bind two aptamers (TBA), Wang et al. constructed a SERS aptasensor based on a sensing interface with a sandwich type system of TBA/α-thrombin/TBA–Au nanoparticles (Fig. 13). It was shown that the Raman signal of the reporters attached to the AuNPs can significantly be enhanced by depositing silver nanoparticles (AgNPs) on the AuNP surface. In fact, the sensors showed a detection limit of 0.5 nM for α-thrombin along with a high selectivity for α-thrombin over other proteases.115
 | ||
| Fig. 13 Schematic illustration of the fabrication process of a SERS aptasensor for protein recognition.115 | ||
Similarly, Cho et al. developed a TBA-based SERRS sensor with a detection limit for α-thrombin of 100 pM.114 The sensing mechanism is based on a single step binding event of α-thrombin to the aptamer, which leads to a decrease of the SERRS signal of the reporter molecule attached to TBA. The decrease in the SERRS signal of the reporter probe is caused by the displacement of TBA from the gold nanoparticle surface, to which it is adsorbed in its unfolded state in the absence of α-thrombin. In the presence of α-thrombin, TBA undergoes a conformational change, leading to TBA being displaced from the gold NP surface and resulting in a reduction in the SERRS signal of the TBA attached Raman tag.114
The concept behind the two discussed aptasensors could feasibly be used to design aptamer based SERS/SERRS platforms for multiple protein detection by using different Raman reporters.
4.2 Protease sensing with magnetic nanoparticles
Magnetic nanoparticles are particularly attractive as core materials when there is a requirement to monitor the distribution of the sample after injecting intravenously or via an extravascular route as magnetic nanoparticles could be non-invasively traced by in vivo MRI-techniques.1Magnetic nanoparticles that serve both as magnetic resonance (MR) contrast agents and NIRF optical probes have been explored by Josephson et al.125 Specifically, a clinically approved superparamagnetic iron oxide particle was used as a template to which protease-degradable fluorophore-labelled peptide substrates were anchored.
A 12-fold increase in the fluorescent signal was observed upon cleavage of the spacer peptidein vitro because of the dequenching of the NIR fluorophore. In vivo, the probe acted both as an MR agent providing information on the location, and when activated by a protease as a NIRF reporter, providing information on the molecular environment.125 One problem associated with this probe was that quantification of protease concentration was not possible as the absolute value of the fluorescence obtained was dependent on the intensity of incident light and the depth and size of the lesion.
An improvement was subsequently reported that used dual fluorophore enzyme activated probes featuring one NIRF fluorochrome that is activated by protease activity and a second fluorochrome that is protease resistant and thus serves as an internal standard.73 While the absolute values of the fluorescence of the two fluorochromes were dependent on the lesion size and the distance of lesions from the surface, the fluorescence ratio was not affected by lesion size and depth, thus allowing improved quantification of enzyme activity.
Inspired by the biological motif of initiating assembly by enzymatic removal of inhibitors, Harris et al. designed a protease triggered nanoparticle self-assembly probe. The binding of biotin and neutravidin-coated superparamagnetic iron oxide nanoparticles was inhibited by the attachment of PEG chains that were linked to MMP-2 cleavable peptide substrates onto the particles. Upon proteolytic removal of PEG, through peptide cleavage, biotin and neutravidin particles self-assembled into nanoassemblies leading to enhanced magnetic susceptibility, transverse (T2) magnetic resonance relaxation and lowered diffusivity.126
4.3 Protease sensing with polymeric and silica-based nanoparticles
Polymers are important classes of materials that have been widely used for the fabrication of nanosized materials for use in bioimaging and biodiagnostics.127,128In 2006 Kim et al. reported apoptosis sensitive NIR-fluorescence-activated polymeric nanoparticles. The probes were produced by conjugating a caspase-3 cleavable substrate to cell permeable polymeric nanoparticles prepared from a hydrophilic polymer (branched polyethyleneimine) and a hydrophobic moiety (deoxycholic acid). In the absence of the target protease, the NIR fluorescence of the fluorochrome Cy5.5 was quenched due to the close spatial proximity of the dye molecules. However, a noticeable increase in the NIR fluorescence signal was observed both in vitro and in vivo in the presence of the caspase-3.129
Another example demonstrating the use of organic fluorescent nanoparticles for in vivoprotease imaging was introduced by McIntyre and co-workers.130 The group designed a polyamidoamine dendrimer-based fluorogenic substrate to image tumour associated matrix-metalloprotease-7 (MMP-7) in vivo. An MMP-7 cleavable peptide labelled with fluorescein (F1) and an internal standard were covalently linked to the dendrimetric scaffold. The internal standard of tetramethylrhodamine (TMR) detected both the cleaved and uncleaved reagents. The two species could be visualised and optically discriminated based on the ratio of the green/red fluorescence (F1/TMR). In vivo measurements showed that the polymeric probe gave significantly enhanced F1 fluorescence from MMP-7 positive but not control tumours.130
Welser et al.87 recently synthesized a new family of fluorescence-based protease responsive nanoprobes (PRNs) based on poly(acrylamide-co-N-(3-aminopropyl)-methacrylamide) nanoparticles (Fig. 14). Green fluorescent bifunctional coumarin fluorophores were attached to both amine functionalised nanoparticles and a peptide substrate that was specific for the protease of interest, subtilisin. While the intact probe displayed minimal absorbance and emission, substantial signal amplification was obtained upon proteolysis, thus allowing an efficient detection of protease activity.
 | ||
| Fig. 14 Model of a protease responsive polymeric nanoprobe.87 | ||
However, it was observed that these amine-bearing nanoparticles aggregate over time, resulting in limited shelf-lives.131 The endeavour to further improve the probes led to the design of azido- and alkyne-modified nanoparticles which were easily transformed into protease responsive nanoprobes by the Huisgen Cu(I)-catalyzed reaction (CuAAC) with click-readied fluorogenic substrates (Fig. 15).131 One of the advantages in this new conjugation strategy is that there is no need for the protection of reactive functional groups in the substrate domain due to the chemoselectivity of the CuAAC reaction.
 | ||
| Fig. 15 Synthetic strategies for the preparation of azido- and alkyne-modified polymeric nanoparticles for the detection of proteases.131 | ||
Unlike some polymer-based nanoparticles, silica derived nanoparticles are robust and highly stable under various thermal and chemical conditions.3 The silica shell of these particles facilitates a wide variety of surface reactions, thus allowing facile conjugation with biomolecules.39 A FRET-based protease biosensor utilising silica nanobeads (15 nm in diameter) was reported by Grant and co-workers.132 The group used trypsin as a model protease and showed that FRET substrates, bearing the trypsin sensitive peptide sequence PRG, were successfully cleaved by the target protease, when attached to silica nanoparticles. Using this probe design, it was possible to achieve a limit of detection of 12.3 µg ml−1.132
5. Summary and outlook
Diverse arrays of nanoparticulate systems are available for protease sensing ranging from inorganic nanoparticles to silica and polymeric based particles. All the available systems have their own advantages and disadvantages and there are concerns about the feasibility of in vivo use for many of these systems due to potential toxicity. However, it has been demonstrated that these probes offer highly sensitive, spatial and quantitative detection and monitoring of protease activity both in vitro and in vivo. It is therefore most likely that their importance in this field will continue to grow, especially as continued developments are being made in manufacturing methods, in the discovery of new biocompatible materials, and in the synthesis of improved imaging reporters. Disease diagnosis and treatment are dependent on an in-depth understanding of biochemical processes and emerging technologies such as nanoparticle based sensing platforms will inevitably play an important role in future advances. With further improvements, the highlighted nanoparticulate systems will become increasingly accepted tools in disease screening, monitoring and the efficient development of more efficacious drugs against diseases where protease activity can be beneficially altered.References
- D. K. Kim and J. Dobson, J. Mater. Chem., 2009, 19, 6294–6307 RSC.
- V. Mailander and K. Landfester, Biomacromolecules, 2009, 10, 2379–2400 CrossRef.
- M. Liong, S. Angelos, E. Choi, K. Patel, J. F. Stoddart and J. I. Zink, J. Mater. Chem., 2009, 19, 6251–6257 RSC.
- C. Fang and M. Q. Zhang, J. Mater. Chem., 2009, 19, 6258–6266 RSC.
- C. M. Byrd and D. E. Hruby, Drug Dev. Res., 2006, 67, 501–510 CrossRef CAS.
- J. T. A. Hsu, H. C. Wang, G. W. Chen and S. R. Shih, Curr. Pharm. Des., 2006, 12, 1301–1314 CrossRef CAS.
- A. G. Tomasselli and R. L. Heinrikson, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1477, 189–214 Search PubMed.
- R. Roy, J. Yang and M. A. Moses, J. Clin. Oncol., 2009, 27, 5287–5297 CrossRef CAS.
- M. Cudic and G. B. Fields, Curr. Protein Pept. Sci., 2009, 10, 297–307 CrossRef CAS.
- D. Leung, G. Abbenante and D. P. Fairlie, J. Med. Chem., 2000, 43, 305–341 CrossRef CAS.
- B. Zhang, Biochem. Biophys. Res. Commun., 2004, 323, 674–678 CrossRef CAS.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, Freeman, W.H., 5th edn, 2002 Search PubMed.
- B. Turk, Nat. Rev. Drug Discovery, 2006, 5, 785–799 CrossRef CAS.
- A. Ingram, L. Byers, K. Faulds, B. D. Moore and D. Graham, J. Am. Chem. Soc., 2008, 130, 11846–11847 CrossRef CAS.
- C. Guarise, L. Pasquato, V. De Filippis and P. Scrimin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3978–3982 CrossRef CAS.
- X. S. Puente, L. M. Sanchez, C. M. Overall and C. Lopez-Otin, Nat. Rev. Genet., 2003, 4, 544–558 CrossRef CAS.
- K. Lin, R. B. Perni, A. D. Kwong and C. Lin, Antimicrob. Agents Chemother., 2006, 50, 1813–1822 CrossRef CAS.
- A. Spaltenstein, W. M. Kamierski, J. F. Miller and V. Samano, Curr. Top. Med. Chem. (Sharjah, United Arab Emirates), 2005, 5, 1589–1607 CrossRef CAS.
- B. Law, R. Weissleder and C. H. Tung, Bioconjugate Chem., 2007, 18, 1701–1704 CrossRef CAS.
- D. R. Edwards and G. Murphy, Nature, 1998, 394, 527–528 CrossRef CAS.
- A. M. Cataldo and R. A. Nixon, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 3861–3865 CrossRef CAS.
- G. Blum, S. R. Mullins, K. Keren, M. Fonovic, C. Jedeszko, M. J. Rice, B. F. Sloane and M. Bogyo, Nat. Chem. Biol., 2005, 1, 203–209 CrossRef CAS.
- G. Blum, G. von Degenfeld, M. J. Merchant, H. M. Blau and M. Bogyo, Nat. Chem. Biol., 2007, 3, 668–677 CrossRef CAS.
- M. Funovics, R. Weissleder and C. H. Tung, Anal. Bioanal. Chem., 2003, 377, 956–963 CrossRef CAS.
- C. Southan, Drug Discovery Today, 2001, 6, 681–688 CrossRef CAS.
- C. M. Salisbury, D. J. Maly and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 14868–14870 CrossRef CAS.
- B. Bresnihan, OMERACT IV Conference, Cancun, Mexico, 1998 Search PubMed.
- S. Chakraborti, M. Mandal, S. Das, A. Mandal and T. Chakraborti, Mol. Cell. Biochem., 2003, 253, 269–285 CrossRef CAS.
- M. G. Paulick and M. Bogyo, Curr. Opin. Genet. Dev., 2008, 18, 97–106 CrossRef CAS.
- L. J. McCawley and L. M. Matrisian, Mol. Med. Today, 2000, 6, 149–156 CrossRef CAS.
- S. C. Dixon, K. B. Knopf and W. D. Figg, Pharmacol. Rev., 2001, 53, 73–91 CAS.
- A. Philchenkov, M. Zavelevich, T. J. Kroczak and M. Los, Exp. Oncol., 2004, 26, 82–97 Search PubMed.
- B. A. Frosch, I. Berquin, M. R. Emmert-Buck, K. Moin and B. F. Sloane, APMIS, 1999, 107, 28–37 Search PubMed.
- T. M. Maguire, S. G. Shering, C. M. Duggan, E. W. McDermott, N. J. O'Higgins and M. J. Duffy, Int. J. Biol. Markers, 1998, 13, 139–144 Search PubMed.
- H. Rochefort, M. Garcia, M. Glondu, V. Laurent, E. Liaudet, J. M. Rey and P. Roger, Clin. Chim. Acta, 2000, 291, 157–170 CrossRef CAS.
- C. H. Tung, Biopolymers, 2004, 76, 391–403 CrossRef CAS.
- D. Stockholm, M. Bartoli, G. Sillon, N. Bourg, J. Davoust and I. Richard, J. Mol. Biol., 2005, 346, 215–222 CrossRef CAS.
- M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS.
- D. Knopp, D. P. Tang and R. Niessner, Anal. Chim. Acta, 2009, 647, 14–30 CrossRef CAS.
- A. P. Alivisatos, W. W. Gu and C. Larabell, Annu. Rev. Biomed. Eng., 2005, 7, 55–76 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- S. Kim, Y. T. Lim, E. G. Soltesz, A. M. De Grand, J. Lee, A. Nakayama, J. A. Parker, T. Mihaljevic, R. G. Laurence, D. M. Dor, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2004, 22, 93–97 CrossRef.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- R. E. Wrolstad, A. D. Decker, S. J. Schwartz and P. Sporns, Handbook of Food Analytical Chemistry, Water, Proteins, Enzymes, Lipids and Carbohydrates, John Wiley & Sons, New Jersey, 2005 Search PubMed.
- T. J. Fan, L. H. Han, R. S. Cong and J. Liang, Acta Biochim. Biophys. Sin., 2005, 37, 719–727 CrossRef CAS.
- V. Gurtu, S. R. Kain and G. H. Zhang, Anal. Biochem., 1997, 251, 98–102 CrossRef CAS.
- A. Baruch, D. A. Jeffery and M. Bogyo, Trends Cell Biol., 2004, 14, 29–35 CrossRef CAS.
- W. Y. Zhong and S. J. Benkovic, Anal. Biochem., 1998, 255, 66–73 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, 1999 Search PubMed.
- A. Yaron, A. Carmel and E. Katchalskikatzir, Anal. Biochem., 1979, 95, 228–235 CAS.
- B. Z. Packard, D. D. Toptygin, A. Komoriya and L. Brand, Methods Enzymol., 1997, 278, 15–23 CAS.
- H. Hirata, A. Takahashi, S. Kobayashi, S. Yonehara, H. Sawai, T. Okazaki, K. Yamamoto and M. Sasada, J. Exp. Med., 1998, 187, 587–600 CrossRef CAS.
- A. Komoriya, B. Z. Packard, M. J. Brown, M. L. Wu and P. A. Henkart, J. Exp. Med., 2000, 191, 1819–1828 CrossRef CAS.
- H. Dacres, M. M. Dumancic, I. Horne and S. C. Trowell, International Workshop on GPCRs—from Deorphanisation to Lead Structure Identification, Berlin, Germany, 2006.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS.
- S. Mittoo, L. E. Sundstrom and M. Bradley, Anal. Biochem., 2003, 319, 234–238 CrossRef CAS.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS.
- G. U. Nienhaus, Angew. Chem., Int. Ed., 2008, 47, 8992–8994 CrossRef CAS.
- R. D. Mitra, C. M. Silva and D. C. Youvan, Conference on Fluorescent Proteins and Applications, Palo Alto, CA, 1995.
- R. Heim and R. Y. Tsien, Curr. Biol., 1996, 6, 178–182 CrossRef CAS.
- M. J. Morgan and A. Thorburn, Cell Death Differ., 2001, 8, 38–43 CrossRef CAS.
- P. Tawa, J. Tam, R. Cassady, D. W. Nicholson and S. Xanthoudakis, Cell Death Differ., 2001, 8, 30–37 CrossRef CAS.
- K. Q. Luo, V. C. Yu, Y. M. Pu and D. C. Chang, Biochem. Biophys. Res. Commun., 2001, 283, 1054–1060 CrossRef CAS.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS.
- M. Kohler, S. V. Zaitsev, I. I. Zaitseva, B. Leibiger, I. B. Leibiger, M. Turunen, I. L. Kapelioukh, L. Bakkman, I. B. Appelskog, J. B. de Monvel, G. Imreh and P. O. Berggren, Diabetes, 2003, 52, 2943–2950 CrossRef.
- P. R. Selvin, Nat. Struct. Biol., 2000, 7, 730–734 CrossRef CAS.
- A. Prinz, G. Reither, M. Diskar and C. Schultz, Proteomics, 2008, 8, 1179–1196 CrossRef CAS.
- Z. Y. Xia and J. H. Rao, Curr. Opin. Biotechnol., 2009, 20, 37–44 CrossRef CAS.
- B. Laxman, D. E. Hall, M. S. Bhojani, D. A. Hamstra, T. L. Chenevert, B. D. Ross and A. Rehemtulla, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16551–16555 CrossRef CAS.
- M. A. O'Brien, W. J. Daily, P. E. Hesselberth, R. A. Moravec, M. A. Scurria, D. H. Klaubert, R. F. Bulleit and K. V. Wood, J. Biomol. Screening, 2005, 10, 137–148 CrossRef CAS.
- M. F. Kircher, R. Weissleder and L. Josephson, Bioconjugate Chem., 2004, 15, 242–248 CrossRef CAS.
- S. Lee, K. Park, K. Kim, K. Choi and I. C. Kwon, Chem. Commun., 2008, 4250–4260 RSC.
- C. H. Tung, S. Bredow, U. Mahmood and R. Weissleder, Bioconjugate Chem., 1999, 10, 892–896 CrossRef CAS.
- C. Bremer, S. Bredow, U. Mahmood, R. Weissleder and C. H. Tung, Radiology (Oak Brook, IL, U. S.), 2001, 221, 523–529 Search PubMed.
- W. Pham, Y. D. Choi, R. Weissleder and C. H. Tung, Bioconjugate Chem., 2004, 15, 1403–1407 CrossRef CAS.
- C. H. Tung, R. E. Gerszten, F. A. Jaffer and R. Weissleder, ChemBioChem, 2002, 3, 207–211 CrossRef CAS.
- J. L. Lauer-Fields, H. Nagase and G. B. Fields, J. Biomol. Tech., 2004, 15, 305–316 Search PubMed.
- C. Grierson, D. Miller, P. LaPan and J. Brady, Anal. Biochem., 2010, 404, 232–234 CrossRef CAS.
- S. L. Yu, N. Wang, C. Y. Liou and W. J. Syu, J. Virol. Methods, 1995, 53, 63–73 CrossRef CAS.
- J. R. Crowther, ELISA: Theory and Practice, Humana Press, 1995 Search PubMed.
- M. Fonovic and M. Bogyo, Curr. Pharm. Des., 2007, 13, 253–261 CrossRef CAS.
- J. E. Ghadiali and M. M. Stevens, Adv. Mater., 2008, 20, 4359–4363 CrossRef CAS.
- S. Lee, E. J. Cha, K. Park, S. Y. Lee, J. K. Hong, I. C. Sun, S. Y. Kim, K. Choi, I. C. Kwon, K. Kim and C. H. Ahn, Angew. Chem., Int. Ed., 2008, 47, 2804–2807 CrossRef CAS.
- C. H. Tung, Y. H. Lin, W. K. Moon and R. Weissleder, ChemBioChem, 2002, 3, 784–786 CrossRef CAS.
- K. Welser, J. Grilj, E. Vauthey, J. W. Aylott and W. C. Chan, Chem. Commun., 2009, 671–673 RSC.
- K. H. Su, Q. H. Wei, X. Zhang, J. J. Mock, D. R. Smith and S. Schultz, Nano Lett., 2003, 3, 1087–1090 CrossRef CAS.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS.
- A. Laromaine, L. L. Koh, M. Murugesan, R. V. Ulijn and M. M. Stevens, J. Am. Chem. Soc., 2007, 129, 4156–4157 CrossRef CAS.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365–370 CrossRef CAS.
- E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar, J. Feldmann, S. A. Levi, F. van Veggel, D. N. Reinhoudt, M. Moller and D. I. Gittins, Phys. Rev. Lett., 2002, 89, 4.
- Y. B. Zhong, F. Zeng, J. Chen, S. Z. Wu, C. Hou and Z. Tong, J. Inorg. Organomet. Polym., 2007, 17, 679–685 CrossRef CAS.
- T. L. Jennings, M. P. Singh and G. F. Strouse, J. Am. Chem. Soc., 2006, 128, 5462–5467 CrossRef CAS.
- W. R. Algar, M. Massey and U. J. Krull, TrAC, Trends Anal. Chem., 2009, 28, 292–306 CrossRef CAS.
- E. Dulkeith, M. Ringler, T. A. Klar, J. Feldmann, A. M. Javier and W. J. Parak, Nano Lett., 2005, 5, 585–589 CrossRef CAS.
- T. Pons, I. L. Medintz, K. E. Sapsford, S. Higashiya, A. F. Grimes, D. S. English and H. Mattoussi, Nano Lett., 2007, 7, 3157–3164 CrossRef CAS.
- S. J. Zhen, Y. F. Li, C. Z. Huang and Y. F. Long, Talanta, 2008, 76, 230–232 CrossRef CAS.
- W. B. Cai, A. R. Hsu, Z. B. Li and X. Y. Chen, Nanoscale Res. Lett., 2007, 2, 265–281 CrossRef CAS.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef.
- J. H. Choi, K. H. Chen and M. S. Strano, J. Am. Chem. Soc., 2006, 128, 15584–15585 CrossRef CAS.
- C. Luccardini, A. Yakovlev, S. Gaillard, M. van't Hoff, A. P. Alberola, J. M. Mallet, W. J. Parak, A. Feltz and M. Oheim, J. Biomed. Biotechnol., 2007, vol. 2007, article ID 68963 Search PubMed.
- L. H. Qu, Z. A. Peng and X. G. Peng, Nano Lett., 2001, 1, 333–337 CrossRef CAS.
- V. Biju, S. Mundayoor, R. V. Omkumar, A. Anas and M. Ishikawa, Biotechnol. Adv., 2010, 28, 199–213 CrossRef CAS.
- X. H. Gao, Y. Y. Cui, R. M. Levenson, L. W. K. Chung and S. M. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759–1762 CrossRef CAS.
- H. Q. Yao, Y. Zhang, F. Xiao, Z. Y. Xia and J. H. Rao, Angew. Chem., Int. Ed., 2007, 46, 4346–4349 CrossRef CAS.
- E. Chang, J. S. Miller, J. T. Sun, W. W. Yu, V. L. Colvin, R. Drezek and J. L. West, Biochem. Biophys. Res. Commun., 2005, 334, 1317–1321 CrossRef CAS.
- Y. P. Kim, E. Oh, M. Y. Hong, D. Lee, M. K. Han, H. K. Shon, D. W. Moon, H. S. Kim and T. G. Lee, Anal. Chem., 2006, 78, 1913–1920 CrossRef CAS.
- I. L. Medintz, A. R. Clapp, F. M. Brunel, T. Tiefenbrunn, H. T. Uyeda, E. L. Chang, J. R. Deschamps, P. E. Dawson and H. Mattoussi, Nat. Mater., 2006, 5, 581–589 CrossRef.
- H. Yao, M. K. So and J. Rao, Angew. Chem., Int. Ed., 2007, 46, 7031–7034 CrossRef CAS.
- M. Suzuki, Y. Husimi, H. Komatsu, K. Suzuki and K. T. Douglas, J. Am. Chem. Soc., 2008, 130, 5720–5725 CrossRef CAS.
- H. Cho, B. R. Baker, S. Wachsmann-Hogiu, C. V. Pagba, T. A. Laurence, S. M. Lane, L. P. Lee and J. B. H. Tok, Nano Lett., 2008, 8, 4386–4390 CrossRef CAS.
- Y. Wang, H. Wei, B. Li, W. Ren, S. Guo, S. Dong and E. Wang, Chem. Commun., 2007, 5220–5222 RSC.
- M. K. So, C. J. Xu, A. M. Loening, S. S. Gambhir and J. H. Rao, Nat. Biotechnol., 2006, 24, 339–343 CrossRef CAS.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Chem. Rev., 1999, 99, 2957–2976 CrossRef CAS.
- I. R. Lewis and H. G. M. Edwards, Handbook of Raman Spectroscopy: From the Research Laboratory to the Process Line, Marcel Dekker, 2001 Search PubMed.
- S. M. Nie and S. R. Emery, Science, 1997, 275, 1102–1106 CrossRef CAS.
- S. J. Lee, A. R. Morrill and M. Moskovits, J. Am. Chem. Soc., 2006, 128, 2200–2201 CrossRef CAS.
- J. W. Chen, X. P. Liu, K. J. Feng, Y. Liang, J. H. Jiang, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2008, 24, 66–71 CrossRef CAS.
- Y. E. K. Lee, R. Smith and R. Kopelman, Annu. Rev. Anal. Chem., 2009, 2, 57–76 Search PubMed.
- B. D. Moore, L. Stevenson, A. Watt, S. Flitsch, N. J. Turner, C. Cassidy and D. Graham, Nat. Biotechnol., 2004, 22, 1133–1138 CrossRef CAS.
- G. L. Liu, Y. T. Rosa-Bauza, C. M. Salisbury, C. Craik, J. A. Ellman, F. F. Chen and L. P. Lee, J. Nanosci. Nanotechnol., 2007, 7, 2323–2330 CrossRef CAS.
- L. Josephson, M. F. Kircher, U. Mahmood, Y. Tang and R. Weissleder, Bioconjugate Chem., 2002, 13, 554–560 CrossRef CAS.
- T. J. Harris, G. von Maltzahn, A. M. Derfus, E. Ruoslahti and S. N. Bhatia, Angew. Chem., Int. Ed., 2006, 45, 3161–3165 CrossRef CAS.
- T. Asefa, C. T. Duncan and K. K. Sharma, Analyst, 2009, 134, 1980–1990 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- K. Kim, M. Lee, H. Park, J. H. Kim, S. Kim, H. Chung, K. Choi, I. S. Kim, B. L. Seong and I. C. Kwon, J. Am. Chem. Soc., 2006, 128, 3490–3491 CrossRef CAS.
- J. O. McIntyre, B. Fingleton, K. S. Wells, D. W. Piston, C. C. Lynch, S. Gautam and L. M. Matrisian, Biochem. J., 2004, 377, 617–628 CrossRef CAS.
- K. Welser, M. D. A. Perera, J. W. Aylott and W. C. Chan, Chem. Commun., 2009, 6601–6603 RSC.
- S. A. Grant, C. Weilbaecher and D. Lichlyter, Sens. Actuators, B, 2007, 121, 482–489 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |