How useful is ion mobility mass spectrometry for structural biology? The relationship between protein crystal structures and their collision cross sections in the gas phase
Ewa Jurneczko and Perdita E. Barran*
The School of Chemistry, The University of Edinburgh, Edinburgh, UK. E-mail: perdita.barran@ed.ac.uk; Tel: +44 (0)131 650 7533
First published on 31st August 2010
Abstract
The technique of ion mobility mass spectrometry (IM-MS) has become of increasing interest for rapid analysis of the conformations adopted by biological macromolecules. It is currently used routinely for analysis of explosives and illegal substances in airport and military security. In biophysical research, it can be used to determine the temperature dependent rotationally averaged collision cross section of gas-phase ions of proteins and nucleic acids along with their mass to charge ratios. Nanoelectrospray ionisation allows the gentle transfer of intact biomolecules from solutions in which the native form(s) are present, into the solvent free environment of a mass spectrometer. It is believed by many researchers that the experimental collision cross sections of these molecules should have some relationship to crystal structure coordinates. In this review we outline the different experimental methods that can be used to measure ion mobility; we also describe methods used to calculate collision cross sections from input coordinates. Following this survey of the methodological approaches to IM-MS, we then summarise IM-MS data published to date for some monomeric peptides and small soluble proteins, along with collision cross sections calculated from their crystal structure coordinates. Finally we consider the relationship between experimental gas-phase conformations and those adopted in crystals and give an outlook on the application of IM-MS as a tool for structural biology.
 Ewa Jurneczko | Ewa Jurneczko is a postgraduate student in the School of Chemistry at Edinburgh University where she is undertaking a PhD in biological mass spectrometry sponsored by the BBSRC and Waters. Her current research in the Barran group investigates the effects of different drift gases on protein ions using IM-MS, as well as exploring the use of IM-MS to examine intrinsic disorder in proteins. She received her BSc in Forensic Science from the University of Northumbria in 2009, which involved a year in industry working for Aptuit. |
 Perdita E. Barran | Perdita Barran is currently a Senior Lecturer in Biophysical Chemistry at the University of Edinburgh. She graduated from Manchester University with a degree in Chemistry with Industrial Experience (1994), and from Sussex University with a PhD in Chemical Physics (1998) under the supervision of Professors Tony Stace and Sir Harry Kroto. She worked as a post-doctoral researcher for Tony Stace, before moving to the University of California Santa Barbara to work with Mike Bowers. She was awarded an EPSRC Advanced Research Fellowship in 2002 which allowed her to commence independent research at Edinburgh University in 2003. |
Introduction
The start of the analytical technique termed ‘ion mobility spectrometry’ (IMS) can be traced to the beginnings of the 20th century, when experimental1,2 and theoretical3 physicists became interested in the movement of ions in gases. In these early studies, as in all modern ion mobility methods what is measured is the velocity of ions in a mass spectrometer where they ‘drift’ due to the presence of a weak electric field while experiencing collisions with a buffer gas. Following the experiments of Erikson1 and Bradbury,2 in the 1950's McDaniel4,5 constructed low field drift tubes which bear a great resemblance to the Drift Time IMS mobility instrumentation still used today. In 1965 Kebarle coupled such a drift tube to a mass spectrometer creating the hybrid technique of ion mobility mass spectrometry (IM-MS).6,7 This work prompted the development of different configurations of ion mobility and mass spectrometry instrumentation over the past 40 years, and their application to an ever wider range of analyte species.8 Details of the early parallel development of ion mobility and of mass spectrometry and its applications to large macromolecules can be found in an excellent review by Heck et al.9 which focussed on applications, and one on instrumentation in a comprehensive summary by Hill et al.8 In the past 5 years commercial instrumentation for IM-MS10 has been developed and the technique has become more widely applied as a research tool.Despite differences in the configuration of mobility devices there are some features common to all IM-MS experiments. A typical mobility experiment requires the injection of a pulse of ions into a chamber filled with a known gas at a known pressure across which is applied an electric field. The time taken for the ions to pass through the chamber is measured. Upon injection into the chamber or drift cell, the ions experience an electrostatic force pulling them through the cell; this force is countered by collisions between the ions and the buffer gas. The behaviour of an ion moving through a gas under the influence of an electric field (E) is dependant on its energy which is determined by the ratio of electric field strength to buffer gas number density, E/N.11 At low E/N the ions are said to be in the low field limit, under this regime the ions have low velocities which are independent of the field strength. At higher E/N the ions may align in the field and their motion becomes dependent on the field strength. In the low field limit, the motion of the ions can be described in more simple terms and most ion mobility measurements are therefore performed in the low field limit. Under these conditions we define the low field mobility, (K) as the constant of proportionality between drift velocity, (vd) and electric field, E as shown by eqn (1).
| vd = KE | (1) |
For experiments performed at sufficiently high pressure, and low drift voltages, ions will quickly be thermally equilibrated and reach a constant drift velocity vd which is determined by the magnitude of the electric field (E) and its influence on the charge of the ion as well as by the number of collisions that the ion makes with the buffer gas. Hence, the mobility, K, must contain terms relating to molecular shape, charge and the buffer gas pressure. These latter properties combine to provide the rotationally averaged collision cross section (Ω) for each ion which is critically dependent on the chosen buffer gas as well as on its temperature (see eqn (2) below). Hence determining vd elucidates Ω which can be interpreted as the shape adopted by a given molecular ion under particular gas phase conditions.
To determine vd and hence K and Ω accurately in ion mobility instrumentation, the length of the drift region must be known precisely and then the average arrival times of ions can be recorded at a detector post the drift region. In IM-MS instrumentation, detection will also provide mass resolution and so the arrival time for a given mass to charge ratio is found. Most laboratories will provide K in terms of the reduced mobility K0, normalised for pressure and temperature.
In recent years IM-MS coupled with the soft ionisation techniques electrospray ionisation (ESI) and matrix assisted laser desorption ionistation (MALDI) has gained importance as a tool for the analysis of macromolecules and particularly for its application in determining the conformations adopted by biological molecules.12–16 Efforts in commercial development have concentrated on improving the low duty cycle that can be significant in linear ion mobility experiments, and also on increasing the resolution obtained. Initial studies focused as much on structural measurements as on mixture separation, reflecting the growing potential of mass spectrometry as a tool to provide detailed conformational information on biological moieties.17 The use of IM-MS to study proteins and protein complexes has been particularly rapid. To illustrate this in Fig. 1 we have plotted the number of publications as searched for in the ISI Web of Knowledge database containing the words ‘protein + ion mobility mass spectrometry’ versus the year of their publication.
 | ||
| Fig. 1 Results from a search on ISI Web of Knowledge for the words protein + ion mobility mass spectrometry appearing in title or abstract fields. The number of published articles per year is plotted. The graph starts with the first hit.18 | ||
Of note on this figure are the inflections when Clemmer developed instrumentation in 1997 and then more recently when the Synapt instruments were launched in 2006.
There are three principal types of ion mobility instrumentation that have been sucessfully combined with mass spectrometry. As mentioned above an extensive review of IM-MS instrumentation has recently been provided by Hill and co-authors8 and we urge the interested reader to look there for further details. For the purpose of the current article, the IM component of IM-MS instruments employed for the study of proteins are briefly reviewed, along with a few pertinent examples of their use.
Types of mobility separation
Drift time (DT) IM-MS
Following from the apparatus used by Kebarle, the simplest configuration of IM-MS instrumentation consists of a tube located within a vacuum system filled with a buffer gas of interest across which is applied a weak electric field (5–100 Vcm−1). Ions are steered and injected into a small orifice at one end of the drift tube. They drift due to the field and collisions with the buffer gas until they exit through another small orifice. By measuring the drift velocity of an ion through such a linear drift field and solving eqn (1) above, it is possible to determine its collision cross section (CCS) with some degree of accuracy11 according to the relationship in eqn (2):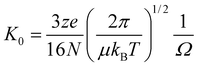 | (2) |
Several IM-MS instruments which incorporate drift cells over which a linear field gradient is applied have been reported. In particular there have been notable efforts made in the ‘modern’ era of IM-MS in designing and building DT IM-MS instrumentation by Bowers with Kemper,20–22 and by Jarrold,23,24 Clemmer,25–31 and Hill.32,33 In a seminal early study, Clemmer and Jarrold used a modified ion mobility-time of flight (TOF) mass spectrometer equipped with an ESI source for analysis of small organic and biological molecules, in particular bradykinin as well as the larger proteinscytochrome c and ubiquitin. Another ESI capable IM-MS instrument IM-Quadrupole reported by Wyttenbach, Kemper and Bowers,22 has been applied to the study of an impressive range of biologically relevant molecules including the amyloidgenic proteins alpha-synuclein16 and the amyloid β protein,34PNA/dsDNA complexes35 and a systematic study of variants of the hormoneGnRH.36 The studies from this group are frequently accompanied by extensive MD simulations of the analytes of interest, allowing comparison between calculated and measured CCSs. The Clemmer group at Indiana University have been responsible for a number of notable advances in instrument and technique development for the study of large proteomic datasets,37,38 including multidimensional ion mobility.25,39–41 In our group we have recently built an ion mobility mass spectrometer, capable of performing temperature variable measurements of CCSs and have applied it to examine the conformations of biological molecules and complexes.42,43 McLean et al. have coupled a DT-IM-MS with imaging mass spectrometry44 - the results show the spatial localization of lipids in biological tissues.
Travelling wave (TW) IM-MS
Waters MS Technologies introduced the first commercially available integrated IM-MS instrument the Synapt HDMS in 2006.10 With its origins in the pioneering work of Gerlich45 this mobility separator consists of three stacked ring ion guides (called the Triwave) with RF applied to consecutive electrodes; to propel ions through the device, a travelling wave (TW) comprising a series of transient DC voltages is superimposed on top of the RF voltage. In 2009, Waters launched the Synapt G2 System which improves some of the design features of the first generation instruments, whilst essentially still a TW mobility separator. This second generation instrument has a higher field pusher into a dual stage reflectron. Together these features provide higher sensitivity, improved mass accuracy, and an elevated data acquisition rate of 20 spectra per second.46 Moreover the ion mobility resolving power of the Synapt G2 is up to four times higher than that of the original Synapt HDS system. In the short time these instruments have been available, they have made a significant impact on the use of IM-MS for the study of macromolecular systems and in particular on the study of intact proteins and protein complexes. (see Fig. 1). The mobility of ions through these travelling wave devices is not as directly related to collision cross section as in DT IM-MS instruments. Shvartsburg has made notable efforts to formally understand the relationship between collision cross section and mobility in a travelling wave device,47 but as yet there is no adoption of such an approach by the IM-MS community. Despite this, with a careful use of standards previously measured in heliumvia DT IM-MS, it is possible to convert mobilities measured with N2 as a drift gas in Synapt instrumentation into mass selected ‘helium based’ collision cross sections.48,49 Using a Synapt HDS system, Robinson et al. have assessed conformations of multimeric proteins,50 and also the disassembly of complexes viewing the partial unfolding of monomer units whilst still retaining some of the integrity of the complex.51 Leary and Robinson have employed IM-MS along with careful controlled collision induced dissociation to map two subunits of the human eukaryotic initiation factor 3 protein. Ashcroft has used TW IM-MS methodology to examine prefibrillar aggregates of amyloidgenic proteins.52 Scrivens has applied Synapt technology to several applications for biomolecular science including a study of the protein haemoglobin,53 and with Bowers a study of the calcium binding protein calmodulin. Both of these studies complement our work using DT IM-MS.54 Heck and co-workers have examined very large species with TW IM-MS including chaperonin complexes55 and intact virus particles.56 Oldham has performed promising experiments which apply IM-MS to the stabilisation of proteins by ligands.57 As can be seen in Fig. 1 the number of publications which employ IM-MS to examine proteins has grown significantly since the introduction of the Synapt instruments by Waters. We have not provided an exhaustive list here, rather a flavour of the range of biological systems amenable to this analysis.Field asymmetric waveform ion mobility spectrometry
The final type of ion mobility separation, commonly used in conjunction with mass spectrometry is field asymmetric waveform ion mobility spectrometry (FAIMS) or differential mobility spectrometry. First developed in the USSR in the early 1980's,58 it was sucessfully coupled with ESI-MS by Purves, Gruevremont and co-workers in the late 1990's.59,60 The FAIMS/MS platform was made commercially available in 2003 as a front-end device that could be coupled to a number of mass spectrometers. In this technique ions pass between two electrodes in the presence of a tangential gas flow. FAIMS devices are usually placed at the source end of mass spectrometers where the gas flow may well be due to the drying gas requirements of an electrospray source. The voltages applied to the electrodes and the speed of the gas flow can be tuned such that ions of a specific mobility K are transmitted successfully through the device. This said, separation in FAIMS is independent of the absolute value of K, ions are separated by the difference in their mobilites at high and low values of E. Ashcroft and co-workers have employed a front end mobility separator available from Thermo-Electron coupled to a quadrupole TOF instrument to examine conformers of β-microglobulin.61 The group of Smith have been extremely successful in both the implementation with respect to separation of complex mixtures62 and the interpretation of FAIMS63 From this group, of particular note is a recent report which discusses the pendular alignment of proteins in gases under the influence of electric fields which has great promise as a new analytical approach for studying protein unfolding and for conformer selection of large systems.64 Cooper has also used FAIMS to help separate proteomic mixtures, with an emphasis on elucidation of phosphopeptides.65 This latter study and the work of Smith et al. probably best represent the strengths and future applications of the FAIMS approach to ion mobility as an analytical technique for separation of mixtures, essentially gas phase chromatography, rather than for precise conformational detail.Calculation of collision cross sections (CCSs)
Because the mobility of a given ion is inversely proportional to its CCS, it follows that mobilities and buffer gas specific CCSs could be accurately calculated for any set of coordinates for subsequent comparison with experimental values. This would involve evaluation of orientationally averaged cross sections with an approximate but appropriate treatment of ion–buffer gas collisions. Indeed this can be accomplished, and Jarrold66–68 and Bowers69 have made significant breakthroughs in this direction. Currently, MOBCAL66,67,68 developed by Shvartsburg and Jarrold is widely used to determine the theoretical CCSs of biological molecules. This open source software program is based on three different treatments of the ion-buffer gas collision which calculate rotationally averaged cross sections from input coordinate files, derived from X-ray crystallography or NMR studies or from MD simulations. With the exception of some recent work by Shvartsburg,70 all computations rely on averaging the orientation of a set of coordinates and calculating the interaction with a chosen buffer gas. The contained models in MOBCAL are: the projection approximation (PA),69 exact hard sphere scattering method (EHSS)66,71 and trajectory method (TM).68The above three methods determine the CCS with helium as the in silicobuffer gas, which to date somewhat curtails the use of other gases in experiments. A particularly simple approach is the projection method (PA)69,66 which has been employed to obtain rotationally averaged cross sections of proposed molecular structures for over 80 years.72 The CCS is determined by averaging the geometric projection areas over all possible orientations. However this method ignores the long-range interactions and all the details of the scattering process between the ion and buffer gas. In order to address the inadequacy of the PA approach, Wyttenbach69 implemented the use of scaled size parameters for all of the atoms in the molecule, however this still does not address the effects of multiple collisions, and hence for ions of masses greater than 2 kDa, or for particularily crenulated conformations of smaller molecules such as those found for crown ethers, this method will underestimate CCSs and is of limited value for larger biomolecules. In the exact hard sphere scattering (EHSS), a CCS is calculated by averaging the momentum transfer cross section which is related to the scattering angles between the incoming and departing gas atom trajectory. This model takes into account scattering and the collision process but does not consider the effects of long range potentials between the buffer gas and the molecular ion.73 The trajectory method (TM)68 is considered to be the most reliable and accurate method for calculating the CCS especially for larger ions. This approach takes into account the long-range interactions and close collisions between the ion and buffer gas atom, as well as the effects of multiple collisions. To perform this calculation, the effective potential must be defined and then the trajectories are run within this potential to obtain the scattering angles.
Ion mobility measurements on monomeric proteins
As discussed above IM-MS has been used to investigate many aspects of biological systems: from separating complex proteomic mixtures74 to determining the conformations of large macromolecular complexes.50 To best consider the benefit and applicability of IM-MS for structural biology, we have decided to examine cross section measurements reported for monomeric proteins of differing mass. We list these proteins in Table 1 along with the CCSs obtained for the lowest charge state observed experimentally, as well as the primary reference. All measurements used electrospray or nanoelectrospray to ionise the proteins, we also detail the solution conditions for each case. It can be seen that the CCSs broadly scale with molecular mass, a point that has been made before.9 Of note is the apparent reproducibility between measurements taken on the same protein in different laboratories: for example we reported 1217 Å2 for the [M + 5H]5+ ion of cytochrome c sprayed from pure water using DT IM-MS54 and Smith et al.75 report 1238 Å2 for the same charge state under buffered conditions using TW IM-MS.| Protein Name | Experimental Method | Solution Condition | Experimental Collision Cross Section (Å2) | Source |
|---|---|---|---|---|
| a The unpublished data can be found on the webpages of the Barran89 and Clemmer90groups.b This value has been estimated from the graph in ref. 12. | ||||
| melittin | DT IM-MS | distilled H2O | 544 (+3) | Florance, Barran et al. (unpublished results)a |
| human beta defensin- 2 | DT IM-MS | distilled H2O | 598 (+3) | De- Cecco, Barran et al. (unpublished results)a |
| bovine pancreatic trypsin inhibitor | DT IM-MS | 75 : 25 H2O/AcN, with acetic acid | 770b (+4) | Shelimov, Clemmer, et al.12 |
| ubiquitin | DT IM-MS | 50 : 50 H2O : AcN, with 2.0% acetic acid | 1004, 1059 (+4) | Valentine, Counterman, et al.86 |
| TW IM-MS | 20 mM ammonium acetate (pH 7) | 791 (+4) | Smith, Knapman et al.75 | |
| beta-2-microglobulin | TW IM-MS | 10 mM ammonium formate (pH 2.6) | 1142 (6+) | Smith, Knapman et al.75 |
| cytochrome c | DT IM-MS | distilled H2O (pH 5.65) | 1217 (+5) | Faull, Korkeila, et al.54 |
| 75 : 25 H2O : AcN with acetic acid | 1139(+3) | Shelimov, Clemmer, et al.12 | ||
| TW IM-MS | 20 mM ammonium acetate (pH 7) | 1238 (+5) | Smith, Knapman et al.75 | |
| alpha-lactalbumin | TW IM-MS | 20 mM ammonium acetate (pH 7) | 1342 (+6) | Smith, Knapman et al.75 |
| lysozyme | DT IM-MS | 50 : 50 H2O : MeOH with 0.1% formic acid | 1300 (+5) | McCullough, Kalapothakis, et al.42 |
| 50 : 50 H2O : AcN with 0.2% acetic acid | 1313 (+5) | Valentine, Anderson, et al.87 | ||
| TW IM-MS | 20 mM ammonium acetate (pH 7) | 1333 (+6) | Smith, Knapman et al.75 | |
| 25 mM ammonium acetate with 10% MeOH (pH 6.9) | 1487 (+8) | Hopper, and Oldham57 | ||
| apo-calmodulin | DT IM-MS | 10 mM ammonium acetate | 1526, 1750 (+7) | Faull, Korkeila, et al.54 |
| 90 : 10 10 mM bicarbonatebuffer : MeOH | 1630, 1900 (+7) | Wyttenbach, Grabenaauer et al.88 | ||
| apo-myoglobin | DTIM-MS | not available | 1459 (+4) | Clemmer (unpublished results)a |
| myoglobin (with heme) | TWIM-MS | 20 mM ammonium acetate (pH 7) | 1314 (+6) | Smith, Knapman et al.75 |
| 25 mM ammonium acetate;10% MeOH (pH 6.9) | 1742 (+8) | Hopper, and Oldham57 | ||
| haemoglobin, tetramer [α2β2H4] | DTIM-MS | 50 mM ammonium acetate (pH 6.75) | 3051 (+13) | Faull, Korkeila, et al.54 |
Calculated collision cross sections for monomeric proteins
We have used all three models implemented in MOBCAL as described above to estimate the theoretical collision cross sections for the monomeric proteins listed in Table 1. We started with published X-ray crystallography measurements (RCSB protein data bank (PDB)) with the exception of apo-calmodulin for which theoretical helium CCSs have been obtained using the NMR coordinates. Since all crystal structures do not possess hydrogen atoms, we have also used AmberTools76 to add hydrogens to all of the amino acids for each coordinate set. We present results for both the raw crystal structure coordinates and for the form with hydrogens added (Table 2). The results presented show that CCSs obtained using the PA method are on average up to 20% smaller than those obtained from the other two approaches (this difference is marginally greater for the larger molecules). As described above, this is because the PA method ignores the effects of multiple scattering events and so we ask the reader to discount the PA values here as they are certain to be inaccurate and misleading for the larger proteins. On the other hand the EHSS model gives values that are slightly higher, especially for smaller molecules, than those determined by TM. It can be seen that as the molecular size increases, the values obtained by EHSS and TM converge. These deviations can be explained by the fact that PA does not take into account the scattering effect, the EHSS model overestimates the cross sections for small ions simply because it (as well as PA) in MOBCAL has been parameterized for fullerenes and other large ions of similar size, and small ions form shallower ion-molecule potentials that are effectively manifested as smaller collision radii, finally the long range interactions included in TM appear to be of less importance for larger ions, but TM properly accounts for the size dependent variation of the interaction potential. For all of the systems the addition of hydrogens, which of course are present in the experimental molecular species, does increase the collision cross sections. This difference lessens with molecular mass, from 3.5% for BTPI to 2% for haemoglobin. These deviations are within experimental error for IM-MS measurements however since the effect of hydrogen addition is always to increase the CCS we do not think that this effect can be ignored.| Protein Name | MW (Da) | PDB | Project Approximation (Å2)a | Exact Hard Sphere Scattering (Å2) | Trajectory Method (Å2) |
|---|---|---|---|---|---|
| a As described in the text, we consider the projection approximation unsuitable for valid comparison with experiment, and are only included here for completeness.b For melittin the crystal structure coordinates are a dimer. We extracted a monomer from this for the calculations for comparison with experiment.c Experimental measurements were made on bovine calmodulin Bos Taurus not on that from Xenopus laevis. The sequence homology between the two species is >99% with a single amino acid difference. The only structure available for the apo form is viaNMR and comes already with resolved hydrogen atoms. | |||||
| honey bee melittinb, monomer | 2846 | 1MLT91 | 493 | 586 | 574 |
| with hydrogens | 520 | 621 | 609 | ||
| human beta defensin- 2 | 4328 | 1FD392 | 575 | 691 | 669 |
| with hydrogens | 601 | 725 | 692 | ||
| bovine pancreatic trypsin inhibitor | 6512 | 6PTI93 | 749 | 914 | 891 |
| with hydrogens | 777 | 952 | 918 | ||
| bovine ubiquitin | 8565 | 1UBQ94 | 885 | 1088 | 1055 |
| with hydrogens | 905 | 1113 | 1088 | ||
| human beta-2-microglobulin | 11860 | 1LDS95 | 1163 | 1454 | 1459 |
| with hydrogens | 1192 | 1488 | 1494 | ||
| equine cytochrome c | 12355 | 1HRC96 | 1056 | 1317 | 1310 |
| with hydrogens | 1085 | 1352 | 1351 | ||
| bovine alpha-lactalbumin | 14178 | 1HFX97 | 1205 | 1511 | 1523 |
| with hydrogens | 1235 | 1548 | 1560 | ||
| chicken lysozyme | 14305 | 1DPX98 | 1172 | 1461 | 1468 |
| with hydrogens | 1196 | 1490 | 1484 | ||
| African Clawed frog calmodulinc (apo-CaM) | 16700 | 1CFD99 | 1652 | 2090 | 2029 |
| sperm whale, myoglobin (with heme) | 17566 | 1VXG100 | 1377 | 1737 | 1725 |
| with hydrogens | 1410 | 1783 | 1776 | ||
| haemoglobin, tetramer [α2β2H4] | 64447 | 1GZX101 | 3217 | 4201 | 4181 |
| with hydrogens | 3268 | 4277 | 4267 |
Comparison of calculated collision cross sections for monomeric proteins with experimental values
In Fig. 2 below, the results of our survey of the literature on experimental IM-MS studies of monomeric proteins (Table 1) is compared with the results of our calculations of the helium CCS from the crystal structure coordinates of the proteins (Table 2). | ||
| Fig. 2 Summary of the data presented in Tables 1 and 2. Here the experimental collision cross section for the lowest observed charge state for each small peptide or protein is plotted along with the value found from calculation for the form of the crystal structure that we have added hydrogen atoms to. The value of the calculated collision cross section is taken from that found via the TM method. We have not included the haemoglobin tetramer in this figure due to its greater molecular mass. | ||
We can see from this figure that for everyprotein the experimental CCS is lower than the value found from crystal structure coordinates. In general this effect is more marked as the size of the protein increases. So for the small haemolytic peptide melittin, the experimental measurement for the [M + 3H]3+ monomeric form of this peptide is 544 Å2 whereas the calculated CCS for the form with hydrogens is 609 Å2 which translates to a difference of just over 10%. Melittin is present as a dimer in the crystal structure data and the monomer from this has been cut out of the pdb file for CCS calculation. The melittin monomer is a ‘banana’ shaped α- helix due to a proline residue in the middle of its sequence. This structure must collapse significantly in the gas phase. The same effect is seen for the largest protein we have considered here, haemoglobin, where the experimental measurement for the CCS of the tetramer is significantly smaller (now by some 28% from the crystal structure coordinates). For haemoglobin this effect can be attributed to a collapse of the structure upon desolvation which might be especially marked because the crystal structure possesses a significant space in the centre of the tetramer. This is consistent with the findings of Matthews that the fraction of the crystal volume occupied by solvent ranged from 27% to 78% for proteins under 70 kDa, although more recently for higher resolution crystallographic data this figure has been revised to a median value of 47%.77 Of course this is based on the unit cell dimensions which are greater or equal to the radius of gyration of the crystal structure coordinates used for the CCS calculations, nonetheless given that the masses of the proteins analysed correspond to fully or near fully desolvated molecular ions, it is clear that loss of solvent has allowed the partial collapse of the structure preserved in the crystalline form. This is most marked when the protein has any void in the middle as for haemoglobin. For these small soluble proteins, such a loss of a stable solvated conformation as found in the crystal structure on entry to the gas phase is not at all surprising. Even with careful nanoeletrospray ionisation the gas-phase is a distinctly different environment than solutions. By assessing experimental and theoretical studies, Breuker and McLafferty have reasonably surmised that following desolvation charged side chains will collapse within picoseconds and in milliseconds (the timescales of most IM-MS experiments) there will be a loss of hydrophobic interactions and a subsequent dissociation of electrostaic bonds.78 For small monomeric proteins there is a strong likelihood that these processes will significantly alter the solvated structure into a gas phase form. Whether or not this form is still functional is most likely protein dependent.
Conclusions and outlook
With small biomolecules molecular dynamics simulations performed in the gas phase can provide an accurate representation of the molecular species observed in an IM-MS experiment36,79–81 Clemmer has also considered the use of intrinsic size parameters to approximate the structures of amino acids in peptides.82 More recently Bernstein et al. have used coarse grained methods to consider the packing of amyloidgenic proteins.83 With larger systems it is also common to compare the change in collision cross section from that obtained from a crystal or NMR structure calculated as we have done here. But invariably for monomeric proteins, the experimental collision cross section for the lowest recordable charge state is always smaller than that obtained from the crystal structure coordinates. Conversely for higher charge states, experimental CCSs are often higher than those given by crystal structure data,12,42,43,54,84,85 which is attributable to coulombically driven unfolding in the gas phase, as well as the transfer of more denatured proteins from solution. These experimental observations are somewhat in conflict with the ‘holy grail’ of native proteinmass spectrometry where we aim to preserve the functional form of a protein into the mass spectrometer for analysis. Retention of conformation, composition and stability of proteins in the gas phase has always been a major concern when dealing with native proteinsassemblies, and indeed there are recent examples of the use of IM-MS to study disruption of proteinassemblies upon transfer into the gas phase.49,51 This critical review serves to emphasize this point. We conclude that care must be taken when interpreting results from IM-MS measurements, and in particular on attempts to correlate measurements with results from crystallography. One sensible approach might be to first perform a gas-phase minimisation on the crystal or NMR coordinates via molecular dynamics prior to calculation of the gas-phase CCS for comparison with gas-phase experimental CCS values: such an approach has been attempted by Shea, Bowers79 and co-workers albeit with simulated solvated structures as their starting point. It would also be interesting to compare gas-phase collision cross sections with structural measurements on proteins made using other techniques for example light scattering or electrophoresis.Measurement of K in DT IM-MS instrumentation is indeed an exact science, and buffer gas and temperature dependent CCSs obtained in this way will be intrinsic to the molecular species under those conditions. Because of this, experimental CCSs determined by IM-MS are certainly valid measurements with particular application to the study of stabilisation of protein structure in the presence of ligands,57 other proteins,49 following mutation43 and in particular to the study of protein unfolding.18 We also believe that IM-MS has a very important role to play when examining for example, amyloidgenic precusors where the proteins will be far more packed than in their native state.52,83 For these globular soluble monomeric proteins IM-MS allows us to measure a very collapsed form of the protein under observation through to more extended denatured forms. The fact that IM-MS provides for isolated biomolecules both mass and charge values as well as information about the gas phase, conformation(s) for each m/z combination should never be ignored. The behaviour of proteins in solution will be more dynamic than in the crystalline state, and IM-MS can provide snapshots of this dynamism as well as provide us with an insight into the intrinsic properties of protein fold.
Acknowledgements
We thank all present and prior members of the Barran group: Sally Shirran, Bryan McCullough, Hayden Eastwood, Hannah Florance, Andrew Stopford, Wutharath Chin, Peter Faull, Jason Kalapothakis, Stefan Esswein, Fiona McAllister, Martin De Cecco, Roland Chu, Yana Berezovskaya, Hattie Cole, and Judith Nicholson, as well as all the many project students and collaborators who have helped to support our studies. We are very grateful for the input of Cait MacPhee, Kathrin Breuker, and Alex Shvartsburg in their critical reading of this manuscript. We also must thank EPRSC, BBSRC, the Royal Society, the School of Chemistry and the BMSS for financial support of our work.Notes and references
- H. A. Erikson, Phys. Rev., 1927, 30, 339–348 CrossRef CAS
.
- N. E. Bradbury, Phys. Rev., 1932, 40, 0508–0523 CrossRef CAS
- P. Langevin, Annales De Chimie Et De Physique, 1903, 28, 433–530 Search PubMed
- E. W. McDaniel, W. S. Barnes and D. W. Martin, Rev. Sci. Instrum., 1962, 33, 2 CrossRef CAS
- W. S. Barnes, D. W. Martin and E. W. McDaniel, Phys. Rev. Lett., 1961, 6, 110 CrossRef CAS
- A. M. Hogg and P. Kebarle, J. Chem. Phys., 1965, 43, 449 CrossRef CAS
- P. Kebarle and A. M. Hogg, J. Chem. Phys., 1965, 42, 668 CrossRef CAS
- A. B. Kanu, P. Dwivedi, M. Tam, L. Matz and H. H. Hill, J. Mass Spectrom., 2008, 43, 1–22 CrossRef CAS
- C. Uetrecht, R. J. Rose, E. van Duijn, K. Lorenzen and A. J. R. Heck, Chem. Soc. Rev., 2010, 39, 1633–1655 RSC
- S. D. Pringle, K. Giles, J. L. Wildgoose, J. P. Williams, S. E. Slade, K. Thalassinos, R. H. Bateman, M. T. Bowers and J. H. Scrivens, Int. J. Mass Spectrom., 2007, 261, 1–12 Search PubMed
- E. A. Mason and E. W. McDaniel, Transport properties of ions in gases, Wiley, New York, 1988 Search PubMed
- K. B. Shelimov, D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1997, 119, 2240–2248 CrossRef CAS
- E. R. Badman, C. S. Hoaglund-Hyzer and D. E. Clemmer, Anal. Chem., 2001, 73, 6000–6007 CrossRef CAS
- S. J. Valentine and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2002, 13, 506–517 CrossRef CAS
- J. Gidden, A. Ferzoco, E. S. Baker and M. T. Bowers, J. Am. Chem. Soc., 2004, 126, 15132–15140 CrossRef CAS
- S. L. Bernstein, D. F. Liu, T. Wyttenbach, M. T. Bowers, J. C. Lee, H. B. Gray and J. R. Winkler, J. Am. Soc. Mass Spectrom., 2004, 15, 1435–1443 CrossRef CAS
- J. L. P. Benesch, B. T. Ruotolo, D. A. Simmons and C. V. Robinson, Chem. Rev., 2007, 107, 3544–3567 CrossRef CAS
- D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1995, 117, 10141–10142 CrossRef CAS
- L. M. Matz, H. H. Hill, L. W. Beegle and I. Kanik, J. Am. Soc. Mass Spectrom., 2002, 13, 300–307 CrossRef CAS
- P. R. Kemper and M. T. Bowers, J. Am. Soc. Mass Spectrom., 1990, 1, 197–207 CrossRef CAS
- P. R. Kemper, N. F. Dupuis and M. T. Bowers, Int. J. Mass Spectrom., 2009, 287, 46–57 Search PubMed
- T. Wyttenbach, P. R. Kemper and M. T. Bowers, Int. J. Mass Spectrom., 2001, 212, 13–23 Search PubMed
- P. Dugourd, R. R. Hudgins, D. E. Clemmer and M. F. Jarrold, Rev. Sci. Instrum., 1997, 68, 1122–1129 CrossRef CAS
- M. F. Jarrold, J. Phys. Chem., 1995, 99, 11–21 CrossRef CAS
- S. I. Merenbloom, S. L. Koeniger, S. J. Valentine, M. D. Plasencia and D. E. Clemmer, Anal. Chem., 2006, 78, 2802–2809 CrossRef CAS
- S. J. Valentine, S. L. Koeniger and D. E. Clemmer, Anal. Chem., 2003, 75, 6202–6208 CrossRef CAS
- S. Myung, Y. J. Lee, M. H. Moon, J. Taraszka, R. Sowell, S. Koeniger, A. E. Hilderbrand, S. J. Valentine, L. Cherbas, P. Cherbas, T. C. Kaufmann, D. F. Miller, Y. Mechref, M. V. Novotny, M. A. Ewing, C. R. Sporleder and D. E. Clemmer, Anal. Chem., 2003, 75, 5137–5145 CrossRef
- C. S. Hoaglund-Hyzer and D. E. Clemmer, Anal. Chem., 2001, 73, 177–184 CrossRef CAS
- C. S. Hoaglund, S. J. Valentine, C. R. Sporleder, J. P. Reilly and D. E. Clemmer, Anal. Chem., 1998, 70, 2236–2242 CrossRef CAS
- Y. S. Liu, S. J. Valentine, A. E. Counterman, C. S. Hoaglund and D. E. Clemmer, Anal. Chem., 1997, 69, A728–A735
- C. S. Hoaglund, S. J. Valentine and D. E. Clemmer, Anal. Chem., 1997, 69, 4156–4161 CrossRef CAS
- X. T. Tang, J. E. Bruce and H. H. Hill, Rapid Commun. Mass Spectrom., 2007, 21, 1115–1122 CrossRef CAS
- D. Wittmer, B. K. Luckenbill, H. H. Hill and Y. H. Chen, Anal. Chem., 1994, 66, 2348–2355 CrossRef CAS
- S. L. Bernstein, T. Wyttenbach, A. Baumketner, J. E. Shea, G. Bitan, D. B. Teplow and M. T. Bowers, J. Am. Chem. Soc., 2005, 127, 2075–2084 CrossRef CAS
- E. S. Baker, J. W. Hong, B. S. Gaylord, G. C. Bazan and M. T. Bowers, J. Am. Chem. Soc., 2006, 128, 8484–8492 CrossRef CAS
- P. E. Barran, R. W. Roeseke, A. J. Pawson, R. Sellar, M. T. Bowers, K. Morgan, Z. L. Lu, M. Tsuda, T. Kusakabe and R. P. Millar, J. Biol. Chem., 2005, 280, 38569–38575 CrossRef CAS
- J. A. Taraszka, A. E. Counterman and D. E. Clemmer, Fresenius J. Anal. Chem., 2001, 369, 234–245 CrossRef CAS
- S. J. Valentine, M. Kulchania, C. A. S. Barnes and D. E. Clemmer, Int. J. Mass Spectrom., 2001, 212, 97–109 Search PubMed
- A. E. Hilderbrand, S. Myung, C. A. S. Barnes and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2003, 14, 1424–1436 CrossRef CAS
- M. H. Moon, S. Myung, M. Plasencia, A. E. Hilderbrand and D. E. Clemmer, J. Proteome Res., 2003, 2, 589–597 CrossRef CAS
- Z. Y. Xun, R. A. Sowell, T. C. Kaufman and D. E. Clemmer, J. Proteome Res., 2007, 6, 348–357 CrossRef CAS
- B. J. McCullough, J. Kalapothakis, H. Eastwood, P. Kemper, D. MacMillan, K. Taylor, J. Dorin and P. E. Barran, Anal. Chem., 2008, 80, 6336–6344 CrossRef CAS
- M. De Cecco, E. S. Seo, D. J. Clarke, B. J. McCullough, K. Taylor, D. Macmillan, J. R. Dorin, D. J. Campopiano and P. E. Barran, J. Phys. Chem. B, 2010, 114, 2312–2318 CrossRef CAS
- J. A. McLean, W. B. Ridenour and R. M. Caprioli, J. Mass Spectrom., 2007, 42, 1099–1105 CrossRef CAS
- D. Gerlich, Adv. Chem. Phys., 1992, 82, 1–176 CAS
- http://waters.com/.
- A. A. Shvartsburg and R. D. Smith, Anal. Chem., 2008, 80, 9689–9699 CrossRef CAS
- B. T. Ruotolo, J. L. Benesch, A. M. Sandercock, S. J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 Search PubMed
- J. A. Leary, M. R. Schenauer, R. Stefanescu, A. Andaya, B. T. Ruotolo, C. V. Robinson, K. Thalassinos, J. H. Scrivens, M. Sokabe and J. W. Hershey, J. Am. Soc. Mass Spectrom., 2009, 20, 1699–1706 CrossRef CAS
- B. T. Ruotolo, K. Giles, I. Campuzano, A. M. Sandercock, R. H. Bateman and C. V. Robinson, Science, 2005, 310, 1658–1661 CrossRef CAS
- B. T. Ruotolo, S. J. Hyung, P. M. Robinson, K. Giles, R. H. Bateman and C. V. Robinson, Angew. Chem., Int. Ed., 2007, 46, 8001–8004 CrossRef CAS
- D. P. Smith, S. E. Radford and A. E. Ashcroft, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6794–6798 CrossRef CAS
- C. A. Scarff, V. J. Patel, K. Thalassinos and J. H. Scrivens, J. Am. Soc. Mass Spectrom., 2009, 20, 625–631 CrossRef CAS
- P. A. Faull, K. E. Korkeila, J. M. Kalapothakis, A. Gray, B. J. McCullough and P. E. Barran, Int. J. Mass Spectrom., 2009, 283, 140–148 Search PubMed
- E. van Duijn, A. Barendregt, S. Synowsky, C. Versluis and A. J. R. Heck, J. Am. Chem. Soc., 2009, 131, 1452–1459 CrossRef CAS
- C. Uetrecht, C. Versluis, N. R. Watts, P. T. Wingfield, A. C. Steven and A. J. R. Heck, Angew. Chem., Int. Ed., 2008, 47, 6247–6251 CrossRef CAS
- J. T. S. Hopper and N. J. Oldham, J. Am. Soc. Mass Spectrom., 2009, 20, 1851–1858 CrossRef CAS
- I. A. Buryakov, E. V. Krylov, E. G. Nazarov and U. K. Rasulev, Int. J. Mass Spectrom. Ion Processes, 1993, 128, 143–148 CrossRef CAS
- R. W. Purves, R. Guevremont, S. Day, C. W. Pipich and M. S. Matyjaszczyk, Rev. Sci. Instrum., 1998, 69, 4094–4105 CrossRef CAS
- D. A. Barnett, B. Ells, R. Guevremont and R. W. Purves, J. Am. Soc. Mass Spectrom., 1999, 10, 1279–1284 CrossRef CAS
- A. J. H. Borysik, P. Read, D. R. Little, R. H. Bateman, S. E. Radford and A. E. Ashcroft, Rapid Commun. Mass Spectrom., 2004, 18, 2229–2234 CrossRef CAS
- K. Tang, F. Li, A. A. Shvartsburg, E. F. Strittmatter and R. D. Smith, Anal. Chem., 2005, 77, 6381–6388 CrossRef CAS
- A. A. Shvartsburg, K. Tang and R. D. Smith, J. Am. Soc. Mass Spectrom., 2005, 16, 2–12 CrossRef CAS
- A. A. Shvartsburg, S. Y. Noskov, R. W. Purves and R. D. Smith, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6495–6500 CrossRef CAS
- Y. Xuan, A. J. Creese, J. A. Horner and H. J. Cooper, Rapid Commun. Mass Spectrom., 2009, 23, 1963–1969 CrossRef CAS
- A. A. Shvartsburg and M. F. Jarrold, Chem. Phys. Lett., 1996, 261, 86–91 CrossRef CAS
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Phys. Chem., 1996, 100, 16082–16086 CrossRef CAS
- A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Chem. Phys., 1998, 108, 2416–2423 CrossRef CAS
- T. Wyttenbach, G. vonHelden, J. J. Batka, D. Carlat and M. T. Bowers, J. Am. Soc. Mass Spectrom., 1997, 8, 275–282 CrossRef CAS
- A. A. Shvartsburg, S. V. Mashkevich, E. S. Baker and R. D. Smith, J. Phys. Chem. A, 2007, 111, 2002–2010 CrossRef CAS
- A. A. Shvartsburg and R. D. Smith, J. Am. Soc. Mass Spectrom., 2008, 19, 1286–1295 CrossRef CAS
- E. Mack, J. Am. Chem. Soc., 1925, 47, 2468–2482 CrossRef
- R. A. Zubarev, D. M. Horn, E. K. Fridriksson, N. L. Kelleher, N. A. Kruger, M. A. Lewis, B. K. Carpenter and F. W. McLafferty, Anal. Chem., 2000, 72, 563–573 CrossRef CAS
- J. A. Taraszka, R. Kurulugama, R. A. Sowell, S. J. Valentine, S. L. Koeniger, R. J. Arnold, D. F. Miller, T. C. Kaufman and D. E. Clemmer, J. Proteome Res., 2005, 4, 1223–1237 CrossRef CAS
- D. P. Smith, T. W. Knapman, I. Campuzano, R. W. Malham, J. T. Berryman, S. E. Radford and A. E. Ashcroft, Eur. J. Mass Spectrom., 2009, 15, 113–130 CrossRef CAS
- http://ambermd.org/.
- K. A. Kantardjieff and B. Rupp, Protein Sci., 2003, 12, 1865–1871 CrossRef CAS
- K. Breuker and F. W. McLafferty, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18145–18152 CrossRef CAS
- A. Baumketner, S. L. Bernstein, T. Wyttenbach, G. Bitan, D. B. Teplow, M. T. Bowers and J. E. Shea, Protein Sci., 2006, 15, 420–428 CrossRef CAS
- Y. Mao, M. A. Ratner and M. F. Jarrold, J. Phys. Chem. B, 1999, 103, 10017–10021 CrossRef CAS
- T. Wyttenbach, J. E. Bushnell and M. T. Bowers, J. Am. Chem. Soc., 1998, 120, 5098–5103 CrossRef CAS
- A. E. Counterman and D. E. Clemmer, J. Am. Chem. Soc., 1999, 121, 4031–4039 CrossRef CAS
- S. L. Bernstein, N. F. Dupuis, N. D. Lazo, T. Wyttenbach, M. M. Condron, G. Bitan, D. B. Teplow, J. E. Shea, B. T. Ruotolo, C. V. Robinson and M. T. Bowers, Nat. Chem., 2009, 1, 326–331 CrossRef CAS
- D. Schroder, H. Schwarz, D. E. Clemmer, Y. M. Chen, P. B. Armentrout, V. I. Baranov and D. K. Bohme, Int. J. Mass Spectrom. Ion Processes, 1997, 161, 175–191 CrossRef
- S. Myung, E. R. Badman, Y. J. Lee and D. E. Clemmer, J. Phys. Chem. A, 2002, 106, 9976–9982 CrossRef CAS
- S. J. Valentine, A. E. Counterman and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 1997, 8, 954–961 CrossRef CAS
- S. J. Valentine, J. G. Anderson, A. D. Ellington and D. E. Clemmer, J. Phys. Chem. B, 1997, 101, 3891–3900 CrossRef
- T. Wyttenbach, M. Grabenauer, K. Thalassinos, J. H. Scrivens and M. T. Bowers, J. Phys. Chem. B, 2010, 114, 437–447 CrossRef CAS
- http://homepages.ed.ac.uk/pbarran/PBRG/docs/Cross%20Sections%20for%20website.pdf.
- http://www.indiana.edu/%7Eclemmer/Research/cross%20section%20database/cs%20database.htm.
- M. Gribskov, D. Eisenberg and L. Wesson, to be published.
- D. M. Hoover, K. R. Rajashankar, R. Blumenthal, A. Puri, J. J. Oppenheim, O. Chertov and J. Lubkowski, J. Biol. Chem., 2000, 275, 32911–32918 CrossRef CAS
- A. Wlodawer, J. Nachman, G. L. Gilliland, W. Gallagher and C. Woodward, J. Mol. Biol., 1987, 198, 469–480 CAS
- S. Vijay-kumar, C. E. Bugg and W. J. Cook, J. Mol. Biol., 1987, 194, 531–544 CrossRef CAS
- C. H. Trinh, D. P. Smith, A. P. Kalverda, S. E. V. Phillips and S. E. Radford, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9771–9776 CrossRef CAS
- G. W. Bushnell, G. V. Louie and G. D. Brayer, J. Mol. Biol., 1990, 214, 585–595 CAS
- A. C. W. Pike, K. Brew and K. R. Acharya, Structure, 1996, 4, 691–703 CrossRef CAS
- M. S. Weiss, G. J. Palm and R. Hilgenfeld, Acta Crystallographica Section D, 2000, 56, 952–958 Search PubMed
- H. Kuboniwa, N. Tjandra, S. Grzesiek, H. Ren, C. B. Klee and A. Bax, Nat. Struct. Biol., 1995, 2, 768–776 CrossRef CAS
- F. Yang and G. N. Phillips Jr, J. Mol. Biol., 1996, 256, 762–774 CrossRef CAS
- M. Paoli, R. Liddington, J. Tame, A. Wilkinson and G. Dodson, J. Mol. Biol., 1996, 256, 775–792 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2011 |