Incorporation of pepsin within zwitterionic, anionic, and cationic lipid monolayers: A comparative study
Tapanendu
Kamilya
b,
Prabir
Pal
a,
Mrityunjoy
Mahato
a,
Ratan
Sarkar
c and
G. B.
Talapatra
*a
aDepartment of Spectroscopy, Indian Association for the Cultivation of Science, Jadavpur, Kolkata, 700 032, India. E-mail: spgbt@iacs.res.in; Fax: +91-33-24732805; Tel: +91-33-24734971
bDepartment of Physics, Narajole Raj College, Narajole, Paschim Medinipur, 721 211, India
cDepartment of Physics, Jogesh Chandra Chaudhury College, 30, Prince Anwar Shah Road, Kolkata, 700 033, India
First published on 10th August 2011
Abstract
The incorporation of water-soluble surface-active enzyme pepsin (PEP) within insoluble zwitterionic 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and anionic stearic acid (SA) monolayer is studied. Furthermore, the results are compared with cationic octadecylamine (ODA). Adsorption of PEP is found to be higher in ODA as compared to that of DPPC and SA. PEP adsorption kinetics in lipids monolayer follows two-step process: diffusion and unfolding. The unfolding of PEP is lower in the case of DPPC than in SA and ODA. π-A isotherm together with high-resolution field emission scanning electron microscope (FE-SEM) images indicate that at higher pressure, PEP molecules tend to squeeze out from the lipids monolayer. PEP forms larger irregular intermolecular aggregates by increment of β-component on SA and ODA monolayer. However, in zwitterionic (DPPC) matrix the α-helix increases with smaller intermolecular aggregates. The overall results specify that zwitterionic (DPPC) monolayer is better choice to obtain protein lipid mixed film than anionic (SA) and cationic (ODA) monolayer.
A. Introduction
Adsorption of proteins/enzymes on biomolecular surfaces is a common event in nature and it has promoted research activities in many scientific fields such as microbiology, biomaterials, pharmacology, or nanotechnology. In particular, the interaction of proteins/enzymes with lipids1 is a complex process, described by the diffusive transport of the protein/enzyme to the lipid surface. The extent of binding depends on a variety of factors such as interactions of protein/enzyme with lipid surface, availability of binding sites, etc. Factors such as complex molecular geometries, presence of multiple competitive species, or possible denaturing of protein/enzyme make it extremely difficult to understand and quantitatively analyze protein/enzyme interactions with lipid surfaces. However, one can study protein–lipid interactions to predict their transport, distribution, accumulation, etc., based on the structure and hydrophilicity/hydrophobicity of protein/enzyme molecules. The immobilization of protein/enzyme onto suitable substrate with minimal denaturation have been found extensively crucial for their biological importance in purification of a variety of drugs, peptides, antibodies as well as applicability in developing various biomolecular devices.1–7 In this regard, varieties of methods have been introduced to immobilize biomaterials onto solid matrix surfaces, such as drop cast, sol–gel process, self-assembly, and Langmuir Blodgett (LB) techniques etc.8–13 Among the numerous proposed methods for immobilizing proteins/enzymes, the LB technique of monolayer transfer on solid surfaces with controlling the film thickness and the molecular organization/architecture is the most versatile and convenient techniques for designing ultra thin films of biologically functional molecules.14–16Usually, for the preparation of a protein/enzyme film by the LB method, the aqueous solution of the protein/enzyme is spread very carefully on the water subphase and then the monolayer is compressed to a desired surface pressure for its successful transfer onto appropriate substrate. This is the elementary process associated with the preparation of thin film of protein/enzyme by LB technique. The main disadvantage of this process is that due to the affinity of protein/enzyme molecules to dissolve into water subphase, the surface pressure and molecular area of the obtained monolayer of protein may decrease with time. As a result, one may not get the actual area/molecule of the monolayer. As the interactions between ions and protein/enzyme in aqueous solutions play an important role, the addition of some amount of salt in the subphase is used for decreasing the quantity of molecules of protein/enzyme that sink into the bulk, resulting in the configuration of more stable and organized Langmuir monolayer.17–21 However, our earlier studies state that the electrolyte increases the unfolding/denaturation of protein/enzyme at air/water interface.22
Therefore, incorporation of protein/enzyme in lipid monolayer is a challenging approach to overcome the above problem. The cationic, zwitterionic, or anionic amphiphilic molecules are used to increase the surface activity and to immobilize proteins/enzymes on suitable substrate, depending on the net charge of the protein/enzyme. The functionality of proteins/enzymes depends mainly on intramolecular reorganization due to specific intermolecular interaction with neighboring molecules. This may cause unfolding, and form irreversible aggregates.8 Thus, the immobilization of protein without denaturation and aggregates on lipid film is extremely important in designing such biomolecular devices. Therefore, a comparative study among the adsorption of a surface-active protein/enzyme within anionic, zwitterionic and cationic lipid monolayer is important to understand not only the possible interaction mechanism, but also to find out a suitable protein–lipid complex and/or thin film with minimum denaturation/unfolding. However, these studies are rather rare.
In our recent communications,23 the interaction of a soluble surface-active enzyme, the pepsin (PEP) with cationic octadecylamine (ODA) is studied at air/water interface. This study creates a question about the behavior and the possible mechanism of association of PEP in zwitterionic and in anionic monolayer. To realize the interaction of PEP with the zwitterionic and anionic monolayer at the air/water interface and compare with cationic lipids, the interaction and the incorporation of PEP within the zwitterionic 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and anionic stearic acid (SA) was studied here.
To understand the physical aspect of PEP-lipids interaction/incorporation of PEP within the DPPC and SA surface, the surface pressure (π) versus time (t) and the surface pressure (π) versus area (A) isotherms were studied. A surface compressibility (β-π) study was also done to characterize the states of the PEP-SA as well as the PEP-DPPC monolayer. The present results are compared with the PEP-ODA24 systems. By combined use of LB deposition, field emission scanning electron microscope (FE-SEM) and Fourier transformed infrared spectroscopy (FTIR) techniques, this study has been focused on the structural organization/conformational regulation and the surface morphology of the enzyme–lipid mixed films.
B. Experimental section
DPPC, SA, and ODA were purchased from Sigma and Aldrich Chemical Co., respectively. PEP was purchased from Lobachemie, India. These chemicals were used as received without further purification. The spectral grade chloroform (SRL, India) was used as solvent to prepare solution of DPPC, SA, and ODA with desired concentrations. A Teflon-bar-barrier type LB trough (model 2000C, Apex Instruments Co. India) was used for the preparation, characterization, and deposition of the monolayer film. The trough width and length are 200 mm and 450 mm respectively. The subphase volume is about 1.2 L. The aqueous subphase consisted of pure triple distilled and deionized water obtained from a Milli-Q water purification system via an ELIX from Millipore (Billerica, MA). The pH and the resistivity of the distilled water were 5.5 and 18.2 MΩ cm respectively. All the experiments were performed at temperature 26 ± 1 °C unless otherwise mentioned.To study the adsorption behavior of PEP at bare air/water interface, PEP was injected into the water subphase from the previously prepared aqueous stock solution to attain the required final concentration (0.1 mg/mL) in subphase of LB trough. Whereas, to study the adsorption kinetics of PEP in lipid monolayer, at first the lipid monolayer was formed on the surface of water subphase by spreading a chloroform solution of the lipid (1 mM) onto the subphase with a micro syringe. 20 min was allowed for the spreading solvent to evaporate, then the films were compressed to the desired surface pressure by applying lateral pressure at a rate of 1 Å2/(molecule.min). The aqueous solution of PEP was then injected into the subphase to attain the required final concentration. By keeping the film area constant, changes in π were monitored with time as the protein diffused into the interfacial region. The details of experimental processes were similar with our previous experiment described elsewhere.23,24
For preparation of pure PEP film, a known amount of aqueous solution of PEP of concentration 1 mg/mL was spread on the water subphase by a micro syringe. A similar technique was used for the preparation of lipid monolayer where chloroform solution of lipid (1 mM) was spread on the water subphase by a micro syringe.
For the π-A isotherm measurement of PEP-lipid system, first we mixed the PEP with subphase with concentration of PEP (CPEP) ranging over 0.0001, 0.0005, 0.001 mg/mL. Then the lipid solution was spread on this PEP containing subphase. In all the cases mentioned above, the films were compressed to the required pressures with a compression speed of 1 Å2/(molecule min), after a delay of 20 min from spreading.
Finally, after two hours of stabilization of surface pressure of deposition, all the films were transferred with a speed of 5 mm/min onto very carefully cleaned hydrophilic glass cover slips and silicon wafers, which were previously immersed in the subphase. The cleaning procedure was reported in our earlier literature.9
The surface morphology of all films was studied by high-resolution field emission scanning electron microscope (FE-SEM, model no JEOL JSM-6700 F).
Apart from the π-t and π-A studies, FTIR spectra of PEP in all films on silicon wafer substrate were recorded at room temperature on Magna-IR (Model No 750 spectrometer, series II), Nicolet, USA. In all the cases, the data were averaged over 100 scans. The resolution of the instrument was 4 cm−1.
C. Results and discussion
C.1. Kinetics of PEP incorporation/association within lipids monolayer
Earlier we reported the results of surface activity, i.e.π-t measurements of pure PEP at various concentrations of PEP along with the association behavior of PEP with ODA Langmuir monolayer.23,24 The CPEP dependent π-t curves showed that there were initial lag times (τlag) where τlag values remained near zero. This initial lag phase is the diffusion-limited regime, a significant characteristic of protein/enzyme adsorption process, where the interface is lacking sufficient quantity of protein/enzyme for noticeable change in π. Thus, the lag time is the time required for attaining the minimum monolayer coverage for an effectual and measurable surface pressure.23–25After the lag phase, an initial faster increase followed by a slower increase of π was observed. The first step is the diffusion of the molecule to just below the interface after which the adsorption is in effect at the air/water interface. In the second step, rearrangement among the adsorbed protein/enzyme molecules takes place, resulting in partial unfolding of the adsorbed segment.23–26 Earlier studies24 also showed that the increase of CPEP decreases τlag and makes the diffusion rate faster. We have found out the optimum/critical concentration, Cc of pepsin (= 0.1 mg/ml) to fill up the surface after which no further penetration takes place.
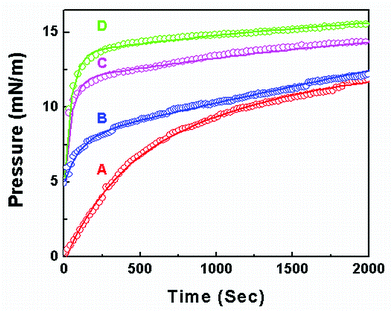
Fig. 1(A) (taken from our earlier study24) shows the changes in π of the PEP solution of CPEP = 0.1 mg/mL injected inside the water subphase. This is a plot of surface pressure contributed by PEP molecules at the air/water interface with time. It shows that in this case the τlag is around 160 s. To visualize clearly the inset of Fig. 1 represents the same up to 700 s. Fig. 1(B–D) represent the change in π of DPPC, SA and ODA monolayer, pre-compressed at the expanded region (π =5 mN/m) monitored with t; respectively, when PEP was injected beneath the lipid monolayer in the water subphase. These curves show that the presence of lipids at the air/water interface alters the shape of the curve. Increase in π immediately after the injection of the PEP solution with vanishing lag time is observed in all cases, implies that the PEP molecules interact and incorporate with the lipid monolayer. However, the equilibria are attained at different surface pressures for different lipids (∼14.2, 14.9, and 16.3 mN/m for DPPC, SA, and ODA respectively) within same duration. Moreover, the rate of incorporation of PEP is faster and most of the PEP uptake occurs in case of ODA than DPPC and SA. In other words, the interaction of PEP-ODA is stronger than PEP-SA and PEP-DPPC. The electrostatic interaction between the head groups of lipids and oppositely charged groups of PEP together with the hydrophobic interaction may be responsible for higher accumulation of the PEP at the air/water interface in presence of lipids. The similar type of observations of increase of surface activity of ovalbumin in presence of the same lipids (DPPC, SA, and ODA) was reported in our earlier literature in detail.27
 | ||
| Fig. 1 Adsorption of PEP (CPEP = 0.1 mg/mL). (A) Bare air/water interface; (B) in DPPC monolayer pre compressed at 5 mN/m; (C) in SA monolayer pre compressed at 5 mN/m; (D) in ODA monolayer pre compressed at 5 mN/m. The inset represents the same up to 700 s. | ||
Fig. 2 represents the time dependent changes in π of the DPPC (Panel A) and SA (Panel B) monolayer pre-compressed at various surface pressures, πpc, when a fixed amount (0.1 mg/mL final concentration) of PEP was injected beneath the lipid monolayer in the water subphase. It is found that Δπ = πt − πpc (πt represents the surface pressure at time t) decreases with the increase of πpc as shown in the panel C and D for DPPC and SA, respectively. At πpc = 30 mN/m, a very small amount of PEP uptake occurs for both in DPPC and SA. In the case of SA, a decrease of π is observed (panel B) due to nucleation and dissolution of SA into subphase at very higher π. At higher πpc, due to small amount of PEP penetration, the change in surface pressure becomes less significant. Thus, after long time π-t plot takes a steady value. This plot of Δπ versus πpc gives the value of the critical pressure (πc) at ∼33.6 and ∼27.1 mN/m for DPPC-PEP and SA-PEP, respectively. πc is the pressure at which the protein cannot produce any further increase of surface pressure. In our earlier work,23 we also found that the Δπ decreases with the increase of πpc, for ODA-PEP system. The critical pressure (πc) was found at ∼31.0 mN/m for ODA-PEP system.
 | ||
| Fig. 2 Panel-A: Adsorption of PEP at various pre compressed surface pressures, (πpc = 5, 10, 18, and 30 mN/m) in DPPC monolayer. Panel-B: Adsorption of PEP at various pre compressed surface pressures, (πpc = 5, 10, 18, 22, and 30 mN/m) in SA monolayer. The panel C and D represent the Δπ vs. πpc plot for DPPC and SA, respectively. | ||
Moreover, the increment of π at the expanded region is more than that at the condensed region, which clearly demonstrates that the PEP penetration behavior is different in the above-mentioned two regions. The protein association or incorporation is easier in the expanded region. The slower adsorption rate of PEP at the interface in presence of condensed lipid film compared to expanded film could be due to the difference in permeability of the respective lipid monolayer. Only a negligible amount of PEP penetrates in condensed lipid films and thus the surface pressure remains unaltered. A slow attachment process is also competing here. Similar observations were found for ovalbumin in DPPC, ODA and SA9,27,28 as well as puroindoline in DPPC and in DPPG monolayer.29
To analyze the kinetics of PEP adsorption, the curves of Fig. 1 are fitted to the following double exponential association mechanism from which some basic features of PEP adsorption can be extracted.23,24,27,28,30 A non-linear least square fitting based on Levenberg-Marquardt algorithm of Microcal Origin 7.5 was used for curve fitting.
 | (1) |
In the case of adsorption at the bare air/water interface, we rejected the initial part belongs to lag time and rescaled the PEP adsorption curve, where the zero time was adjusted when the surface pressure starts to increase.24 The rescaled adsorption curves and their respective fitting curves are presented in the Fig. 3. The parameters resulting from the fits as well as the residual square correlation coefficient (R2) are summarized in Table 1.
 | ||
| Fig. 3 Adsorption kinetics of PEP by fitting eqn (1). (A) Bare air/water interface; (B) in DPPC monolayer pre compressed at 5 mN/m; (C) in SA monolayer pre compressed at 5 mN/m; (D) in ODA monolayer pre compressed at 5 mN/m. Solid lines are fitted curves fitted by eqn (1). | ||
| Lipids | A 1 | t 1 (sec.) | A 2 | t 2 (sec.) | R 2 |
|---|---|---|---|---|---|
| a A1 and A2 are the relative amplitudes and t1 and t2 are corresponding time constants of two mechanisms involving in association process. (R2) is the residual square correlation coefficient. b Data of ODA is taken from our earlier work.24 | |||||
| ODA b | 9.41 ± 0.04 | 37.70 ± 0.83 | 5.84 ± 0.03 | 1350.3 ± 3.49 | 0.999 |
| SA | 6.79 ± 0.01 | 46.80 ± 0.57 | 5.24 ± 0.01 | 1387.4 ± 3.86 | 0.999 |
| DPPC | 2.88 ± 0.02 | 107.43 ± 1.90 | 11.43 ± 0.46 | 4205.5 ± 235.5 | 0.995 |
| No Lipid | 5.62 ± 0.01 | 352.14 ± 4.71 | 8.01 ± 0.08 | 1401.0 ± 18.99 | 0.998 |
The value of t1 decreases more in the presence of lipids than in the bare air/water interface at a particular concentration (0.1 mg/mL) of PEP in the subphase. Moreover, these values imply that diffusion of PEP is slowest in the bare air/water interface and fastest in the presence of the ODA monolayer. The values of the diffusion rate increase as we go from bare air/water interface to presence of SA, DPPC, and ODA monolayer. Maximum uptake of PEP occurs in the presence of ODA monolayer.
The highest value of t2 in the DPPC-PEP system indicates that the amplitude of unfolding and reorganization is less dominant in presence of DPPC. The above results indicate that the DPPC monolayer is far better to increase the surface activity of PEP and for the formation of protein–lipid mixed films.
C.2. π-A isotherm
 | ||
| Fig. 4 π-A isotherm at room temperature (26 °C). (A) Pure DPPC; (B) DPPC + 0.0001 mg/ml PEP; (C) DPP + 0.0005 mg/ml PEP. | ||
The π-A isotherms of PEP mixed DPPC monolayer show that with increasing concentration of PEP in water subphase, the area/molecule of DPPC increases. Probably two processes, one related to the transport/diffusion of PEP at the surface and the other the interaction of PEP with DPPC molecules at the surface control this observation. From Fig. 4, it is distinct that in the condensed region (at π ∼ 50 mN/m), all the π-A isotherms of DPPC-PEP mixed monolayer tend to overlap with the isotherm of pure DPPC. This may indicate successive squeezing out of protein at high surface pressure.9,31,32
 | ||
| Fig. 5 π-A isotherm at room temperature (26 °C). (A) Pure SA; (B) SA + 0.0001 mg/ml PEP; (C) SA + 0.0005 mg/ml PEP. | ||
Fig. 5B and 5C demonstrate the typical π-A compression isotherms of the SA-PEP complex LB monolayer on pure water subphase with various concentrations of PEP, CPEP (0.0001, 0.0005 mg/ml), in water subphase, respectively. It can be seen that the π-A isotherms of the PEP mixed SA monolayer have a sigmoidal28 shape and are quite different from the pure SA monolayer. The area/molecule of the SA monolayer increases with increasing PEP concentration in the water subphase indicate that PEP molecules were incorporated into the SA monolayer from the subphase. The separations among the isotherms with different amount of PEP are more at lower π region. Above π ∼ 40 mN/m, the isotherms approach each other, but they do not overlap i.e. there is no isobestic point common to all the isotherms. In this transition region, most of the PEP molecules are partially squeezed out from the complex PEP-SA monolayer during compression process. Although the squeezing out of PEP at this high surface pressure occurs, yet some of the PEP molecules still attached in the complex monolayer, as there are not a case of complete expulsion of PEP.
Isotherms of ODA-PEP mixed monolayers are discussed in our earlier work.23 In the ODA-PEP mixed monolayer, we found that at higher surface pressure a complete expulsion or successive squeezing out of PEP occurs. At this high surface pressure, the PEP molecules already bound with the ODA may also drag back in to the water subphase along with some of the ODA molecules from the air/water interface due to strong electrostatic interactions.33 However, the isotherm of PEP-DPPC mixed monolayer follows the path of pure DPPC at higher pressure. Therefore, interaction between DPPC and PEP is not strong enough to drag the DPPC at the time of the squeezing out of PEP. In the case of SA, complete expulsion of PEP is not found. These observations indicate that the interaction between SA and PEP is not so strong, but stronger than DPPC and PEP as well as between ODA and PEP.
C.3. FTIR study
Fig. 6A and 6B represent the FTIR spectra of the thin LB film of PEP-DPPC and PEP-SA lifted from the pure water subphase at π =35 mN/m respectively. Unfolding, intra and intermolecular associations of PEP were studied by monitoring the peak positions and widths of two amide bands within a fixed range.34 Both the spectra (A and B) consist of two broad peaks from ∼1500 to 1600 cm−1 and from ∼1600 to 1700 cm−1. The spectrum from 1600 to 1700 cm−1 is the vibration band of amide-I, mainly resulted from carbonyl stretching vibrations of the peptide backbone, sensitive to the peptide secondary structure like α-helices, β-sheets, or β-turns. The vibrational frequency of each C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond depends on the strength of the hydrogen bond and the interactions between the amide units, both of them are influenced by the secondary structure.34,35 The peak from 1500 to 1600 cm−1 can be ascribed to the vibration band of amide-II resulting from a combination of N–H in-plane bending and C–N stretching vibrations of the peptide groups.34,35 The vibrational energies of the carboxyl group depend on the different conformations of the protein, such as α-helix, β-sheet, β-turns, random coil structure, intra and intermolecular aggregates. The determination and the assignment of the spectral components of the amide-I band can then provide information on the secondary structure of the protein.34,36,37 The deconvolution of the normalized amide-I band is useful to study the conformations of the protein/enzyme. A multiple peak fitting technique with Gaussian profile using Micro Cal Origin 7.510,11 was employed to fit a normalized amide-I band to study the conformations of PEP. This multi-component fitting of the profile allows one to identify different components and in particular to determine the corresponding peak frequencies.38 The percentage area of the deconvoluted peaks gives the relative amount of components. It is worth noting that in all the spectra considered in the present work, the maximum number of the components (N), which can be identified in the deconvoluted amide-I band, are 5 (for pure PEP, PEP-DPPC and PEP-SA) as well as 6 (for PEP-ODA)23 to have a meaningful fitting.
O bond depends on the strength of the hydrogen bond and the interactions between the amide units, both of them are influenced by the secondary structure.34,35 The peak from 1500 to 1600 cm−1 can be ascribed to the vibration band of amide-II resulting from a combination of N–H in-plane bending and C–N stretching vibrations of the peptide groups.34,35 The vibrational energies of the carboxyl group depend on the different conformations of the protein, such as α-helix, β-sheet, β-turns, random coil structure, intra and intermolecular aggregates. The determination and the assignment of the spectral components of the amide-I band can then provide information on the secondary structure of the protein.34,36,37 The deconvolution of the normalized amide-I band is useful to study the conformations of the protein/enzyme. A multiple peak fitting technique with Gaussian profile using Micro Cal Origin 7.510,11 was employed to fit a normalized amide-I band to study the conformations of PEP. This multi-component fitting of the profile allows one to identify different components and in particular to determine the corresponding peak frequencies.38 The percentage area of the deconvoluted peaks gives the relative amount of components. It is worth noting that in all the spectra considered in the present work, the maximum number of the components (N), which can be identified in the deconvoluted amide-I band, are 5 (for pure PEP, PEP-DPPC and PEP-SA) as well as 6 (for PEP-ODA)23 to have a meaningful fitting.
 | ||
| Fig. 6 FTIR spectra of LB film of (A) DPPC-PEP; (B) SA-PEP. Inset of (A) and (B) represent the deconvolution of amide band of A and B, respectively. | ||
Neither with the use of fewer number of peaks, the fitting gives a satisfactory result, nor more number of peaks improves the R2 values, and neither do the deconvoluted peaks have definitive positions. We have targeted different peak values for different components to fit the band. The targeting frequencies for β-sheet mode (β-component) and for α-helix mode (α-component) are 1638 cm−1 and 1654 cm−1 respectively. Since the secondary structures are stabilized by hydrogen bonds between amide CO and N–H groups, the position of the components depends on the patterns and the strength of the hydrogen bonds. For stronger hydrogen bonding, the vibration is observed at higher wave number. The β- sheet structures have the strongest hydrogen bonds, exhibit an amide I maximum at much lower frequency than the α-helices.35 The 1618 cm−1 and 1683 cm−1 bands are assigned to inter (A1 component) and intra-molecular aggregates (A2 component) respectively. In the case of the aggregates, the hydrogen bonds formed between the CO and N–H groups of any polypeptides strands with which they come in contact. The consequence is that many hydrogen bonds are formed between polypeptide chains in neighboring protein molecules, forming an aggregate stabilized by very strong intermolecular hydrogen bonds.35 The 1666 cm−1 component can be ascribed to the vibration modes originated by β-turns in the structures (T-component).34,35,38–43 The 1645 cm−1 band is assigned to random coil (RC).44
The inset of Fig. 6A and 6B represents the fitting curves of normalized amide-I peak of LB thin films of PEP-DPPC and PEP-SA, respectively and fitting results are reported in Table 2. All the fitting results are compared with pure PEP LB film, lifted from pure water subphase, studied in our earlier work.24 For all the fittings the square of the correlation coefficient, R2 is found to be 0.999. The summary of the fitting result is indicated in Fig. 7A by bar diagram. The β/α ratio for different films are presented in Fig. 7B by bar diagram.
 | ||
| Fig. 7 Bar diagram of results obtained from FTIR data. A represents the percentage of conformers. Here different components are A1 = inter-molecular aggregates, A2 = intra-molecular aggregates, β = β-sheet structure, α = α-helical structure and T = β-turns structures and RC = random coil. B represents the β/α ratio. | ||
The Fig. 7 (A and B) and Table 2 show that the LB film of PEP-SA lifted from pure water subphase is composed of high amount of β-component (23.07%) and relatively small amount of α-component (12.23%). The value of A1 is (9.11%). Since the β/α ratio (1.88) is greater than unity, most of the α-helix may be converted into β-sheet. In addition, large values of A1 and A2 indicate that there are mostly larger inter and intra molecular aggregates. LB film of PEP-DPPC lifted from pure water subphase is composed of high amount of α-component (31 40%) and relatively small amount of β-component (29.30%). Since the β/α ratio (0.94) is less than unity, some amount of the β sheet may be converted into α helix. The LB thin film of PEP-ODA lifted from pure water subphase is also composed of relatively high amount of A1 (25.01%), and β-component (19.90%) and of small amount of α-component (9.34%).23 The β/α ratio (2.131) is greater than LB film of pure PEP lifted from pure water subphase.24 In the case of pure PEP film the values of β/α ratio and A1 are 1.33 and 8.94%. Thus, more amount of α-helix may convert into β-sheet as well as forms intermolecular aggregates in the case of mixed ODA-PEP and ODA-SA films than PEP-DPPC LB film. Lower values of β/α ratio and A1 in SA matrix than ODA proves that the unfolding of PEP is less in the case of the SA matrix than ODA. We have also found a large amount of RC structure (14.74%) in the case of the mixed ODA-PEP film. This component was not found in pure PEP, PEP-SA, and PEP-DPPC LB film. In case of zwitterionic DPPC matrix, lower values A1 and β/α ratio (0.94) indicate that the unfolding of PEP is less. The overall result indicates that the unfolding and larger intermolecular aggregates formation of PEP is larger in the case of ODA matrix than SA and DPPC by measuring the values of A1. The conversion of β-sheet into α-helix45 in DPPC is the main cause behind the formation of a lower amount of aggregates in the DPPC matrix.
| Conformers | Area (%) | Position (cm−1) | FWHM (cm−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pure PEP | DPPC-PEP | SA-PEP | ODA-PEP b | Pure PEP | DPPC-PEP | SA-PEP | Pure PEP | DPPC-PEP | SA-PEP | |
| a Area (%) = 100 represents the total area under curve. FWHM is full width at half maximum of a peak. b Data of ODA-PEP is taken from our earlier work.24 | ||||||||||
| A 1 | 8.94 | 7.41 | 9.11 | 25.01 | 1623.4 | 1603.9 | 1612.6 | 10.61 | 10.59 | 15.28 |
| β | 39.42 | 29.30 | 23.07 | 19.90 | 1637.0 | 1618.6 | 1623.9 | 17.43 | 20.77 | 9.92 |
| α | 29.73 | 31.40 | 12.23 | 9.34 | 1654.5 | 1654.8 | 1664.5 | 15.93 | 30.02 | 17.02 |
| T | 21.56 | 14.40 | 24.05 | 17.21 | 1669.6 | 1672.4 | 1681.2 | 16.64 | 19.83 | 16.85 |
| A 2 | 0.35 | 17.44 | 31.80 | 13.8 | 1684.7 | 1684.2 | 1694.5 | 05.94 | 14.79 | 19.14 |
| β/α | 1.33 | 0.94 | 1.88 | 2.131 | ||||||
C.4. Surface morphology of transferred monolayer
The surface morphology of the transferred LB films was observed by high-resolution FE-SEM. More details descriptions of pure DPPC, SA, ODA, PEP, and PEP-ODA films are reported in our earlier work.9,23,24,27,28 Only the key points are introduced in this text for completeness. The figures of the above mentioned films are included to compare it with other films. The panel A of Fig. 8 shows the surface morphology of the bare glass slide. The panel B shows that surface morphology of the PEP film transferred at π = 35 mN/m. It consists of two layers. The upper layer consists of larger clusters (20–30 nm) formed by squeezing out of PEP molecules.24 | ||
| Fig. 8 FE-SEM images. (A) Bare Glass; (B) PEP; (C) DPPC; (D) DPPC-PEP; (E) SA (F) SA-PEP; (G) ODA; (H) ODA-PEP. | ||
The panels C and D in Fig. 8 represent the surface morphology of pure DPPC and DPPC-PEP monolayer transferred at a condensed state (∼35 mN/m) on a hydrophilic glass substrate, respectively.
Compact, homogeneous and distinct aggregated domains (20–30 nm in diameter) are found in pure DPPC (Panel C) monolayer.9 Apart from this aggregates, we observe many holes of various sizes (10–100 nm in diameter) through out the films.
For DPPC-PEP monolayer (Panel D) at surface pressure of 35 mN/m the surface is rough and globular domains of proteins are peeping out of the surface. Size of these domains is in the range of 17–20 nm. Absence of these domains at low pressure indicates that at condensed region proteins are squeezed out from the DPPC monolayer and attached to the surface of the DPPC membrane.18 Comparing with the PEP molecular dimension mentioned above, on can say that these domains are aggregates of protein molecules. In addition, protein aggregate capped with DPPC may not be ruled out.
In the case of the SA monolayer (Fig. 8(E)), the film is almost compact with a nanometer scale aggregated nano domains with a regular size of 5–10 nm in diameter.19
The SA-PEP monolayer (Fig. 8(F)) shows a rough surface morphology. In the case of the SA-PEP film, some larger aggregates with 20–100 nm are observed at the upper surface of this monolayer. We believe that these are aggregates of proteins, which form due to squeezing out of protein from the mixed monolayer at condensed region (35 mN/m). Protein clusters also tend to amalgamate and forms larger aggregated structure of protein on the SA monolayer surface.
In the case of pure ODA (Fig. 8(G)), the film is almost compact with the aggregated domains of sizes are around 100 nm. Fig. 8(H) shows the surface morphology of the ODA-PEP monolayer at condensed region (35 mN/m) in micrometer scale resolution. Surface is rough and composed of larger clusters. Here, some patches (holes) are observed in the film due to expelling of the ODA from film together with PEP due to strong electrostatic interactions.
The overall result indicates that the size and the amount of aggregates are smaller in case of the DPPC-PEP system than for the SA-PEP and ODA-PEP systems. These results also support the observations of FTIR.
Conclusions
We have comprehensibly studied the incorporation of water soluble enzyme PEP in different lipid membranes like monolayer of zwitterionic DPPC, anionic SA and cationic ODA at air/water interface. Amount and rate of incorporation of PEP in lipid monolayer is found to be head group charge dependent. Adsorption kinetics studies show that PEP incorporation is more in ODA as compared to that of DPPC and SA. PEP adsorption kinetics in lipids monolayer follows two-step processes: These processes have been identified as diffusion of PEP from the water subphase to the lipid membrane at the air/water interface and subsequent unfolding at the interface. π-A isotherm together with FE-SEM images indicate that at higher pressure, PEP molecules tend to squeeze out from the lipids monolayer. FTIR study of amide bands of transferred monolayers show the β/α ratio of PEP secondary structure increases in the order DPPC < SA < ODA. This implies that the unfolding is less in the case of DPPC than SA and ODA. PEP forms larger irregular intermolecular aggregates by in SA and ODA monolayer due to this unfolding. However, in a zwitterionic (DPPC) matrix the α-helix increases with smaller intermolecular aggregates. The overall results specify that a zwitterionic (DPPC) monolayer is a better choice to obtain protein lipid mixed film than anionic (SA) and cationic (ODA) monolayers.Acknowledgements
We thank DST, Government of India (Project No.-SR/S2/CMP-0051/2006) for partial financial support. Mrityunjoy Mahato thanks CSIR, Government of India for providing the CSIR-NET fellowship.References
- A. Girard-Egrot, P. S. Godoy and L. Blum, Adv. Colloid Interface Sci., 2005, 116, 205 CrossRef CAS.
- F. Beigi and P. Lundahl, J. Chromatogr., A, 1999, 852, 313 CrossRef CAS.
- Y. Okahata, T. Tsuruta, K. Ijiro and K. Ariga, Langmuir, 1988, 4, 1373 CrossRef CAS.
- J. J. Ramsden, Biosens. Bioelectron., 1998, 13, 593 CrossRef CAS.
- S. Hou, J. Wang and C. R. Martin, Nano Lett., 2005, 5, 231 CrossRef CAS.
- Z. Siwy, L. Trofin, P. Kohli, L. A.Baker, C. Trautmann and C. R. Martin, J. Am. Chem. Soc., 2005, 127, 5000 CrossRef CAS.
- C. Leéger and P. Bertrand, Chem. Rev., 2008, 108, 2379 CrossRef.
- J. B. Lee, D. Kim, J. Choi and K. Koo, Colloids Surf., B, 2005, 41, 163 CrossRef CAS.
- T. Kamilya, P. Pal and G. B. Talapatra, J. Phys. Chem. B, 2007, 111, 1199 CrossRef CAS.
- T. Kamilya, P. Pal, M. Mahato and G. B. Talapatra, J. Nanosci. Nanotechnol., 2009, 9, 2956 CrossRef CAS.
- T. Kamilya, P. Pal, M. Mahato and G. B. Talapatra, Mater. Sci. Eng., C, 2009, 29, 1480 CrossRef CAS.
- K. Ariga, J. P. Hill, M. V. Lee, A. Vinu, R. Charvet and S. Acharya, Sci. Technol. Adv. Mater., 2008, 9, 014109 CrossRef.
- P. Pal, D. Nandi and T. N. Misra, Thin Solid Films, 1994, 239, 138 CrossRef CAS.
- V. Rosilio, M. M. Boissonnade, J. Zhang, L. Jiang and A. Baszkin, Langmuir, 1997, 13, 4669 CrossRef CAS.
- P. Pal, M. Mahato, T. Kamilya and G. B. Talapatra, Phys. Chem. Chem. Phys., 2011, 13, 9385 RSC.
- R. Sarkar, P. Pal, M. Mahato, T. Kamilya, A. Chaudhuri and G. B. Talapatra, Colloids Surf., B, 2010, 79, 384 CrossRef CAS.
- A. Ahluwalia, D. De Rossi and J. Shirone, Biosens. Bioelectron., 1991, 6, 133 CrossRef.
- I. Turko, I. Yurkevich and V. Chashchin, Thin Solid Films, 1991, 205, 113 CrossRef CAS.
- L. Dziri, K. Puppala and R. M. Leblenc, J. Colloid Interface Sci., 1997, 194, 37 CrossRef CAS.
- G. K. Chudinova, A. P. Pokrovaskaya and A. P. Savitskii, Russ. Chem. Bull., 1995, 44, 1958 CrossRef.
- V. M. Bolanos-Garcia, S. Ramos, R. Castillo, J. Xicohtencatl-Cortes and J. Mas-Oliva, J. Phys. Chem. B, 2001, 105, 5757 CrossRef CAS.
- T. Kamilya, P. Pal, M. Mahato and G. B. Talapatra, J. Phys. Chem. B, 2009, 113, 5128 CrossRef CAS.
- T. Kamilya, P. Pal and G. B. Talapatra, Biophys. Chem., 2010, 146, 85 CrossRef CAS.
- P. Pal, T. Kamilya, M. Mahato and G. B. Talapatra, Colloids Surf., B, 2009, 73, 122 CrossRef CAS.
- C. Ybert and J. M. Di Meglio, Langmuir, 1998, 14, 471 CrossRef CAS.
- S. C. Biswas, L. Dubreil and D. Marion, J. Colloid Interface Sci., 2001, 244, 245 CrossRef CAS.
- T. Kamilya, P. Pal and G. B. Talapatra, J. Colloid Int. Sci., 2007, 111, 1199 CAS.
- T. Kamilya, P. Pal and G. B. Talapatra, Colloid Surf. B: Biointerfaces, 2007, 111, 1199 CAS.
- L. Dubreil, V. Vie, S. Beaufils, D. Marion and A. Renault, Biophys. J., 2003, 85, 2650 CrossRef CAS.
- J. S. Sharp, J. A. Forrest and R. A. L. Jones, Biochemistry, 2002, 41, 15810 CrossRef CAS.
- E. Polverini, S. Arisi, P. Cavatorta, T. Berzina, L. Cristofolini, A. Fasano, P. Riccio and M. P. Fontana, Langmuir, 2003, 19, 872 CrossRef CAS.
- S. Korl, M. Ross, M. Sieber, S. Kunneke, H. J. Galla and A. Janshoff, Biophys. J., 2000, 79, 904 CrossRef.
- D. Lair, S. Alexandre and J. M. Valleton, Colloids Surf., B, 2005, 45, 200 CrossRef CAS.
- M. Carbonaro, P. Maselli, P. Dore and A. Nucara, Food Chem., 2008, 108, 361 CrossRef CAS.
- Protein–Ligand Interactions: structure and spectroscopy; A Practical Approach Edited by: S. E. Harding, B. Z. Chowdhry, Oxford University Press. Oxford, New York. 2001. Search PubMed.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95 CrossRef CAS.
- Selected applications of modern FT-IR techniques, K. Nishikida, E. Nishio, R. W. Hannah, Gordon and Breach publishers(Tokyo), pp. 268. Search PubMed.
- C. Bhattacharjee, S. Saha, A. Biswas, M. Kundu, L. Ghosh and K. P. Das, Protein J., 2005, 24, 27 CrossRef CAS.
- G. T. Meng and C. Y. Ma, Int. J. Biol. Macromol., 2001, 29, 287 CrossRef CAS.
- J. Bandekar, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1992, 1120, 123 CrossRef CAS.
- K. Murayama and M. Tomida, Biochemistry, 2004, 43, 11526 CrossRef CAS.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073 CrossRef CAS.
- M. Jackson and H. H. Mantsch, Can. J. Chem., 1991, 69, 1639 CrossRef CAS.
- L. Sawer and C. Halt, J. Dairy Sci., 1993, 76, 3062 CrossRef.
- J. Li, Y. Du, P. Boullanger and L. Jiang, Thin Solid Films, 1999, 352, 213 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |