Sorption and spontaneous ionization of phenothiazine within channel type zeolites: Effect of the confinement on the electron transfers.†
Florence
Luchez
,
Sonia
Carré
,
Alain
Moissette
* and
Olivier
Poizat
Laboratoire de Spectrochimie Infrarouge et Raman UMR-CNRS 8516, Bât. C5 Université de Lille 1, 59655, Villeneuve d’Ascq cedex, France
First published on 9th August 2011
Abstract
Diffuse reflectance UV-visible absorption and Raman scattering experimental data show evidence of the phenothiazine (PTZ) sorption and spontaneous ionization in the straight channels of three medium pore acid zeolites with various topologies (ferrierite (H-FER), H-ZSM-5 and mordenite (H-MOR)) but analogous Si/Al contents. The spectral data highlight the combined effects of confinement and local electrostatic field on the sorption and charge separation kinetics. The PTZ incorporation and ionization appeared to be quicker in the larger pore H-MOR than in H-ZSM-5 and in H-FER. However, sorption and ionization are almost complete in the three zeolites after about one year. The low ionization potential value of PTZ (I.P. = 6.73 eV) induced quasi instantaneous formation of the radical cation PTZ•+ in high yield within the internal space of each channel structure. Nevertheless, the higher confinement effect and higher polarizing effect offered by the 10-membered rings (10-MR) channels of H-FER favoured the PTZ second ionization to form the dication PTZ2+. The very long lifetimes of these charge separated states are probably due to the restricted mobility of PTZ in the narrow channels and to the compartmentalization of the trapped electron away from the initial site of PTZ ionization. However, a very slow charge recombination process is observed within the three zeolite morphologies after about one year. This reaction is only partial in the narrower pores of H-FER and H-ZSM-5 whereas the faster diffusion process within the larger pore H-MOR induces quasi total cation disappearance after 2 years. Therefore, the reaction mechanism indicates clearly that PTZ•+ and PTZ2+ are only intermediates and that the thermodynamically stable end product is the occluded PTZ molecule.
Introduction
Comprehensive and detailed characterization of charge transfer processes at interfaces is often delicate to achieve because of the surface heterogeneity and also because of the very short time scales corresponding to formation and recombination of the transient species. These intermediates are generally very reactive and difficult to isolate over long periods. Most electron transfer reactions reported in the literature on porous clays,1,2 oxide and silica gel surfaces,3–6 interfaces like organic and polymeric semi-conductors,7 or in donor–bridge–acceptor molecules8 were initiated through photonic excitation and often required time resolved spectroscopy to be characterized. In that context, zeolites have also shown evidence for their ability to accept electrons and stabilize charge separated states after photoionization of molecules occluded within their cages or channels.9–20 Moreover, it should be noted that in some cases, spontaneous ionization can occur after the mixing of electron donor molecules and channel type zeolites.17,21–23 The mere exposure of molecules having relatively low ionization potential to dehydrated medium pore zeolites was found to induce spontaneous radical cation formation and electron ejection, provided that the pore opening diameter and inner spaces are large enough to allow the molecule incorporation. Note that Bhan and Iglesia demonstrate clearly that the reactivity within acid zeolites is drastically controlled by the pore topology.24It is now well established that the capacity of the zeolite inner space to ionize spontaneously electron donor polyaromatic molecules is essentially correlated to the potential of ionization of the guest molecule as well as to the ionizing ability of the host.23,25 It should be noted that spontaneous ionization was already observed in low yield within non acidic zeolites.26 However, in the case of acidic zeolites, the intrinsic properties of Brønsted acid sites make these host lattices very efficient in ionizing the guest molecules and were found to induce very long lived charge separated states.27,28 The exceptional increase of the transient species lifetime in these systems is assumed to be due to the long distance between the radical cation and the ejected electron. In fact, the latter is trapped far away from the initial ionization site and so from the radical cation.29 Then, both the presence of electron capture sites on the internal zeolite surface and the restricted mobility of the occluded molecule are put forward to explain why back electron transfer reactions are hampered within the narrow zeolite channels. The zeolite internal space also prevents intermolecular reactions from occurring.30
Note that upon heating above 773 K under O2 or inert gas, electron traps can be created within the zeolite framework that can induce electron abstraction from the adsorbed organic molecule even if its ionization potential is high.31,32 In contrast, prior thermal treatment of the zeolite material under inert atmosphere up to 723 K leads only to a release of water and does not create significantly supplementary Lewis acid sites.33 For low ionization potential molecules thermal preactivation is not required.
In previous works, we have already studied spontaneous electron transfer processes within acidic zeolites using various molecules to investigate and make emerge the effects of the guest potential ionization value, of the molecular dimension and shape,25 of the thermal treatment34 and of the Si/T ratio (T = Al or Ga).35,36 Among these works, we have also described the mechanisms of spontaneous electron transfers observed after the exposure of phenothiazine (PTZ) to dehydrated acid ZSM-5 zeolites.37 PTZ has the appropriate dimensions to pass through the pore opening of the ZSM-5 channels and to diffuse in the pores. On the other hand, its low ionization potential value (I.P. = 6.73 eV) makes its ionization very easy. Note that the electron donor properties of PTZ make this molecule and its derivatives highly interesting compounds for optoelectronic applications38,39 and can be very useful in the biological and pharmacological fields.40
In the present work, we have used in situ diffuse reflectance UV-visible absorption (DRUVv) and Raman spectroscopy to monitor the sorption of the rod-shaped phenothiazine molecule in Brønsted acidic ferrierite (H-FER), ZSM-5 (H-ZSM-5) and mordenite (H-MOR) over extended periods of time. The direct exposure of PTZ crystals to the dehydrated zeolites was carried out in the complete absence of solvent and under inert gas. The choice of these three medium pore zeolites with similar Si/Al ratio (∼10) was motivated by the fact that they present analogous channel type structures but differ by their various pore opening diameters. This study was thus aiming at pointing out the consequences of spatial constraints and Brønsted acid site location within zeolite channels on the spontaneous ionization and on the stabilization of charge separated states. The combined effects of confinement and local electrostatic field on the sorption and charge separation kinetics were analyzed using multivariate chemometric methods applied on the experimental spectra. This data processing approach is essential to resolve the diffuse reflection UV-visible absorption spectra of the various individual species involved in the sorption course and to determine their respective spectral concentrations. It should be noted that the above zeolites are commonly used as acid catalysts to carry out hydrocarbon conversion41,42 and that the effects of spatial constraints and acid sites location within these three topologies were already recently investigated for the alkane conversion.43
Experimental
Materials
As-synthesized ZSM-5 samples (Si/Al = 13.5, average particle size ∼1 μm) were obtained according to the template procedure in alkaline medium from VAW aluminum (Schwandorf, Germany). Ferrierite (FER; Si/Al = 10) and mordenite (MOR; Si/Al = 10) were obtained from Zeolyst International (USA). The NH4+ counterbalancing cations of the aluminated zeolites were completely converted to H+ cations by calcination procedure in flowing air by increasing the temperature up to 723 K and holding for 6 h. Phenothiazine (PTZ, C12H9NS, Fluka) was purified by sublimation. Pure and dry Ar gas was used.The unit cell composition of dehydrated H-ZSM-5, H-FER and H-MOR zeolites was found to be H6.6(AlO2)6.6(SiO2)89.4 , H3.3(AlO2)3.3(SiO2)32.7 and H4.3(AlO2)4.3(SiO2)43.7, respectively. However, the27Al MAS NMR experiments carried out on hydrated H-ZSM-5, H-FER and H-MOR show the presence of small quantities of extraframework hexacoordinated Al species.
PTZ Sorption Procedure
Weighed amounts of dehydrated zeolite powder were introduced into an evacuable silica cell under dry Ar and heated to 723 K by successive controlled temperature increases. Then, the samples were cooled to room temperature under dry Ar and weighed amounts of PTZ powder were introduced without any solvent into the cell still under inert atmosphere. The powder mixtures were transferred under dry argon in a quartz glass Suprasil cell and left at 313 K in the dark for long time. The final PTZ loadings at equilibrium correspond to 0.93 PTZ per Unit Cell (UC) for FER, 0.96 PTZ per UC for MOR and 1 PTZ per UC for ZSM-5.Instrumentation
| F(R) = (1−R)2/2R = K/Sc | (1) |
Multivariate Curve Resolution by Alternating Least Squares (MCR-ALS)
The Multivariate Curve Resolution by Alternating Least Squares performs a bilinear decomposition under constraints of multivariate data matrix D(t,λ) according to eqn (2)| D = C . ST + E | (2) |
The process aims at minimizing the residues between the original matrix D and the reconstructed matrix D* obtained by multiplication of the spectral concentration profiles by the pure spectra.
This algorithm enables to provide a description of the kinetic process without any a priori chemical or physical knowledge of the data matrix. Usual constraints for decomposition of spectroscopic data are for instance the non-negativity of the spectra and the concentration or the unimodality of the concentration profiles.
MCR-ALS is an iterative method that needs the initial estimation of either the spectral concentration profiles C or the spectra S. In the present case, the initial estimation of S was obtained by using the SIMPle-to-use Interactive Self-modeling Mixture Analysis (SIMPLISMA) approach.44,45 This algorithm allows the selection of so-called pure variables from the data matrix D. A pure variable is a variable to which one compound of the mixture contributes. All calculations are developed with Matlab 7.7 from the Mathworks. MCR-ALS is performed with the freely available Matlab toolbox developed by Tauler (http://www.vb.es/gesq/mcr/mcr.htm).46
Results
1. Sorption of phenothiazine in H-FER
 | ||
| Fig. 1 Diffuse reflectance UV-visible absorption (DRUVv) spectra recorded after the mixing of solid PTZ and H-FER dehydrated at 723 K under argon. (A) 24 h time window. (B) 27 month time window. (a) 24 h; (b) 3 months; (c) 10 months; (d) 25 months and (e) 27 months after the mixing. Spectra d and e are vertically shifted for clarity. | ||
DRUVv spectra recorded for several months after mixing PTZ and H-FER were analyzed by the MCR-ALS chemometric approach in order to resolve the pure absorption spectra as well as the spectral concentration profiles of each individual species. In the present case, the extraction of four species by MCR-ALS procedure allowed to describe the data matrix. The four pure spectra and their spectral concentration profiles are shown in Fig. 2. Note that these four resolved spectra (Fig. 2A) are very similar to those extracted from the data set obtained after mixing PTZ and the H3.4ZSM-5 zeolite.37 The first spectrum (a) shows bands at 222, 266 and 276 nm. This spectrum is assigned to bulk solid PTZ. The corresponding spectral concentration profile shows a continuous decrease from the mixing (t=0) and suggests the disappearance of solid PTZ in favor of several occluded PTZ species (Fig. 2B, curve a). The second spectrum (b) displays two bands at 250 and 314 nm and is attributed to PTZ adsorbed on the zeolite surface by analogy with the spectra obtained for PTZ after sorption in dealuminated silicalite-1,37 and in methanol solution.47 The third spectrum (c) exhibits a well resolved vibrational structure with band maxima at 440, 475, 497, 512, 754 and 835 nm. This spectrum is assigned to the radical cation PTZ•+ trapped in the porous framework of the zeolite (PTZ•+@H-FER•−). It is consistent with that observed for PTZ•+ stabilized within H3.4ZSM-537 and also formed in sulfuric acid or in acetonitrile solution.48–51 The last species (d) shows two bands in the visible spectral domain at 437 and between 600 and 700 nm. This spectrum is assigned to the dication PTZ2+ because of its similarity with the spectrum of this dication produced by photoinduced electron transfer in phenothiazine/semiconductor systems52 or in sulfuric acid solution.49 Thus, the observation of the PTZ•+ and PTZ2+ spectral features provides evidence of PTZ spontaneous ionization in the porous void space of H-FER. The spectral signature of the ejected electron was not resolved. In addition, the fact that each resolved species exhibits contributions in the UV domain inducing strong overlapping makes difficult the resolution and the exploitation of this spectral range.
 | ||
| Fig. 2 (A) UV-visible absorption spectra of pure species and (B) spectral concentration associated profiles resolved by a MCR-ALS chemometric analysis of the spectra recorded during several months after the mixing of solid PTZ and dehydrated H-FER. (a) (♦) PTZ solid; (b) (▼) PTZ@H-FER; (c) (▾) PTZ•+@H-FER•−; (d) (■) PTZ2+@H-FER2−. | ||
Examining the concentration profiles and the reconstructed D* matrix (not shown) obtained by the MCR-ALS treatment (Fig. 2B) shows that the PTZ•+ formation (curve c) occurs instantaneously with the decay of the solid PTZ (curve a). In contrast, the growth of the occluded PTZ population (curve b) is extremely weak and does not parallel the decay of the solid PTZ. These observations indicate clearly that most of the population of PTZ is directly ionized upon inclusion in the zeolite channels. It can be deduced that the rate of intrazeolitic ionization of PTZ is much faster than the rate of insertion, in such a way that both processes appear quasi simultaneous. Moreover, curve b shows that a weak proportion of the PTZ molecules are also primarily adsorbed in the neutral form. These molecules might be adsorbed initially on the external surface due to the narrowness of the H-FER pore entry which limits the diffusion into the void space. After 2 years, the spectral contribution of this species increases slightly (Fig. 2B).
The PTZ2+ formation (curve d) is slightly delayed with respect to that of PTZ•+ and seems to begin only 20 h after the mixing. Nevertheless, PTZ2+ spectral concentration increased continuously with time and becomes predominant species after about fifty days. The PTZ2+ and PTZ•+ spectral concentrations appear to reach plateaus after about 200 days. However, note that for times longer than 2 years, these spectral concentrations decrease somewhat in favour of the amount of occluded species but PTZ2+ and PTZ•+ are still clearly observed. The relative PTZ•+ and PTZ2+ spectral concentration profiles are illustrated by the high intensity of the PTZ2+ band at 660 nm with respect to the band of PTZ•+ centered at 512 nm in the DRUVv spectra (Fig. 1B). It should be noted that the MCR-ALS approach give only the relative spectral concentration profiles for the four extracted species during the system advancement. Unfortunately, in the absence of molecular extinction coefficient for PTZ2+, relative concentrations of the species can’t be determined precisely. Nevertheless, the experimental data provide evidence of the spontaneous ionization and subsequent dication formation after mixing solid PTZ and H-FER. Due to the PTZ high vapor pressure, sorption occurs in the gas phase and the molecules are assumed to ionize quasi instantaneously. According to the kinetic data, the second ionization is assumed to be slightly delayed with respect to the radical cation formation. Therefore, the reaction process could be described according to the following mechanism:
| PTZgas + H-FER → PTZ@H-FER (sorption) | (3) |
| PTZ@H-FER → PTZ•+@H-FER•− (spontaneous ionization) | (4) |
| PTZ•+@H-FER•− → PTZ2+@H-FER2− (dication formation) | (5) |
| PTZ•+@H-FER•− → PTZ@H-FER (recombination) | (6) |
| PTZ2+@H-FER2− → PTZ@H-FER (recombination) | (7) |
2. Sorption of phenothiazine in H-ZSM5
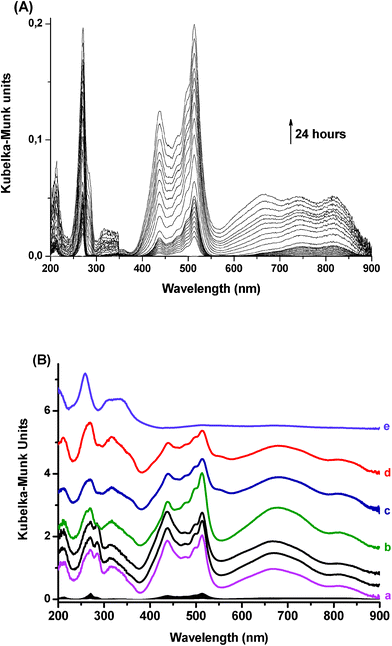 | ||
| Fig. 3 Diffuse reflectance UV-visible absorption (DRUVv) spectra recorded after the mixing of solid PTZ and H6.6ZSM-5 dehydrated at 723 K under argon. (A) 24 h time window. (B) 24 month time window. (a) 8 days; (b) 10 months; (c) 20 months; (d) 24 months after the mixing (e) DRUVv spectra recorded after the mixing of solid PTZ and silicalite-1 dehydrated at 723 K under argon. The spectra are vertically shifted for clarity. | ||
 | ||
| Fig. 4 Spectral concentration profiles resolved by the MCR-ALS chemometric method after the mixing of solid PTZ and dehydrated H6.6-ZSM-5. (a) (♦) solid PTZ; (b) (▼) PTZ@H-ZSM-5; (c) (▾) PTZ•+@H-ZSM-5•−; (d) (■) PTZ2+@H-ZSM-52−. | ||
The decay of the solid PTZ concentration to about zero (Fig. 4, curve a) demonstrates that the PTZ sorption process into H-ZSM-5 is nearly completed after about 100 days. As in H-FER, the concentration profile of adsorbed PTZ (curve b) indicates a very weak amount of neutral molecule within H-ZSM-5. In fact, as can be seen in curves c and d of Fig. 4, the population of occluded PTZ is dominantly present in the cation radical or dication forms, the appearance of which extends over about 100 days, as the decay of the initial solid PTZ. The formation of PTZ•+ and PTZ2+ is thus quasi concomitant with the sorption of PTZ in the zeolite framework. The PTZ•+ concentration profile (curve c) shows clearly that most of the PTZ molecules ionized nearly instantaneously when entering into the pores. The radical cation spectral features at 439 and 513 nm are clearly observed immediately after the mixing (Fig. 3A). Note also that the kinetic of PTZ•+ formation is much faster within H6.6ZSM-5 than within H-FER. The second PTZ ionization appears also to be very rapid as shown by curve (d) profile. The PTZ2+ characteristic band centered at ca. 660 nm is already observed 10 h after the mixing. The spectral concentration profile of PTZ•+ remained always higher than that of PTZ2+ over the studied time scale. The concentrations of these both species reach a plateau after about 100 days and remain very high for at least 1.5 year. Then, the PTZ•+ and PTZ2+ concentrations decrease slightly relatively to the contributions of occluded PTZ observed in the UV domain. To illustrate this feature, the spectrum of PTZ occluded in silicalite-1 in which ionization is weak is presented in Fig. 3B (spectrum e) as a reference. Unfortunately, in the absence of absorbance coefficient value for the dication, no quantitative data can be obtained.
The PTZ ionization and subsequent charge stabilization in H-ZSM-5 was previously characterized using EPR technique.37 The 2D-HYSCORE experiments exhibited a proton pattern resulting from the proton distribution of the PTZ•+ and a very weak coupling between 27Al and the ejected electron. However, we have also shown that, unfortunately, PTZ2+ cannot contribute to the EPR spectrum because of its diamagnetic structure and therefore the EPR data do not provide a correct and representative picture of the system. Nevertheless, analogous long lived charge separated states were also observed after sorption and ionization of other polyaromatics in similar zeolites. The corresponding pulse EPR data suggest that the electron can be trapped as AlO4H•− entities.28
3. Sorption of phenothiazine in H-MOR
 | ||
| Fig. 5 Diffuse reflectance UV-visible absorption (DRUVv) spectra recorded after the mixing of solid PTZ and H-MOR dehydrated at 723 K under argon. (A) 24 h time window, (B) 26 month time window. (a) 24 h; (b) 2 days; (c) 2 months; (d) 3 months; (e) 6 months; (f) 21 months and (g) 26 months after the mixing. Spectra d, e, f and g are vertically shifted for clarity. | ||
The spectral concentration profiles describing the system advancement are presented in Fig. 6. The rapid decrease of the spectral concentration profile corresponding to solid PTZ confirms this assignment and shows that the sorption process into H-MOR is slightly quicker than in H-ZSM-5 and much faster than in H-FER (Fig. 6, curve a). Bulk PTZ was not observed any more after about eighty days. The profiles corresponding to PTZ•+ and PTZ2+ (curves c and d) provide evidence of the very rapid kinetics of the ionization reactions and indicate that the concentration of these ionized species increased for about fifty days. Curves (c) and (d) show that PTZ•+ remains manifestly the major species over the whole investigated time scale. Moreover, the total disappearance of solid PTZ and the appearance of only PTZ•+ and PTZ2+ lead us to assume the total ionization of PTZ within the 12-MR channels of H-MOR. After reaching a maximum after about 2 months, the PTZ•+ and PTZ2+ concentrations seem to decrease slightly for several months. After two years, the characteristic bands of PTZ•+ and PTZ2+ are very weak and the spectrum exhibit mainly 2 bands centered at 315 nm and 250 nm. These bands are assumed to correspond to the spectral signature of occluded PTZ within H-MOR. Therefore, these results clearly confirm the trend previously observed for H-FER and H-ZSM5 showing the recombination of the ionized species with the ejected electrons and resulting in the formation of the thermodynamically stable occluded PTZ.
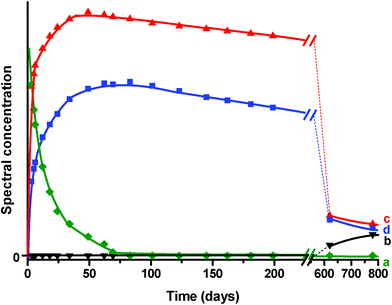 | ||
| Fig. 6 Spectral concentration profiles resolved by the MCR-ALS chemometric method after the mixing of solid PTZ and dehydrated H-MOR. (a) (♦) PTZ solid, (b) (▾) PTZ•+@H-MOR•−, (c) (■) PTZ2+@H-MOR2−. | ||
 | ||
| Fig. 7 FT-Raman spectra (λex = 1064 nm) recorded for solid PTZ (a) and at different times after the mixing of solid PTZ and dehydrated H-MOR. (a) solid PTZ, (b) 1 day, (c) 1 month, (d) 2 months. The resonance Raman spectrum recorded after 2 months (λex = 632.8 nm) is also shown (e). | ||
The weak reappearance of the 1033 cm−1 band after two months after mixing (Fig. 7, spectrum d) is in agreement with the existence of the very slow recombination process suggested above from the observation of a slight decay of the PTZ•+ and PTZ2+ absorption spectra at long time. Unfortunately, high luminescence effects made the monitoring of FT-Raman spectra impossible for times longer than 2 months.
Discussion
The molecular modeling data obtained using Monte Carlo simulation and subsequent energy minimization predict, through the identification of the PTZ sorption sites within the straight 10-MR channels of FER and ZSM-5 and of 12-MR channels of MOR that PTZ sorption probably occurs into these three zeolites. Note that the calculations give only a picture of occluded PTZ within the zeolite channels and do not provide any information on the charge separated states. However, the spectral data obtained during the reaction advancement show the extremely slow disappearance of PTZ•+ and PTZ2+ and let us assume that the theoretical data provide a reasonable picture of the thermodynamically stable end product.Moreover, the UV-visible and Raman spectra demonstrate that fast spontaneous ionization by radical ion formation takes place in high yield within the zeolite void space after the mixing of PTZ crystals and dehydrated zeolite whatever the zeolite topology. Nevertheless, the experimental data show clearly that the charge separated state advancement, namely the second PTZ oxidation and dication formation depends drastically on the pore opening diameter. These different behaviors are discussed below. Note that protonation was never observed upon PTZ sorption into the channels of the three Brønsted acid zeolites despite the acidic strength of the Si-O(H)-Al sites.
Spontaneous ionization
The diffuse reflectance UV visible spectra recorded after mixing PTZ with each one of the investigated zeolites exhibit quasi instantaneously new bands in the visible spectral region between 400 and 550 nm corresponding to PTZ•+. These features evolve gradually for approximately one year to intense absorption bands for the three zeolites before slightly decreasing. However, the kinetics of the ionization process appears to depend on the pore opening size and to decrease markedly from mordenite to ferrierite.The spontaneous ionization efficiency was shown to depend on both the ionization potential value of the occluded molecule and on the ionizing ability of the zeolite internal structure.25,26 The polarization energy at a sorption site is intimately associated with the strength of the local electrostatic field that is particularly high within aluminum rich zeolites because the charge compensating cations located in the channels are only partially shielded.57 Nevertheless, the formation of radical cation at the pore entry and on the external surface can also be assumed if the surface is polarizing enough. However, stabilization of high amount of radical cation is closely associated with the confinement effect. These features were particularly highlighted after mixing anthracene or 9,10-dimethylanthracene and H-ZSM-5. These two molecules present similar ionization potential but anthracene can penetrate into the zeolite channels and show high ionization rate whereas 9,10-dimethylanthracene that cannot enter the pores show very low ionization.25 Recently, the sorption and spontaneous ionization of p-terphenyl (P.I. = 7.8 eV) within the acidic channel types H-FER, H-ZSM-5 and H-MOR simply dehydrated up to 723 K under argon was demonstrated and characterized using spectroscopic tools.58 For molecules with relatively high ionization potential values (>7.5 eV), the ionization rate in zeolites is directly correlated to the aluminium content.34,35 The polarizing effect of the Al-O−(H+)-Si groups within the channels accessible to the guest molecule can generate spontaneous ionization. Then in such cases, accessibility to these acid sites is a key parameter that must be taken into account in the study of ionization. For ferrierite which possesses 10-membered rings (10-MR) and 8-membered rings channels, adsorption techniques show that only the 10-MR channels are accessible to rod-shaped molecules and that these channels contain approximately 40% of the acid sites.59 The pyridine adsorption measurements carried out with ZSM-5 showed that all the acid sites are present in the 10-MR straight and zigzag channels. Then, assuming a random distribution of acid sites between both types of channels, it is reasonable to estimate that at least 60% of the acid sites are situated in the straight channels and/or at the intersection with a sinusoidal channel and thus accessible to rod-shaped molecule like phenothiazine. For mordenite, the distribution of the OH groups is about 40% within the 12-MR channels and 60% in the non-accessible 8-MR channels.60 Thus, according to these considerations, the high ionization rate often observed inside ZSM-5 is not readily comparable with the rates observed within the 10-MR channels of ferrierite or the 12-MR of mordenite even if the total quantity of acid and polarizing sites is similar in all materials (Si/Al ≈ 10). In the present case, experimental data show that PTZ ionization is quasi total within ZSM-5 and mordenite and relatively high within ferrierite, which denotes a lack of specific influence of the zeolite framework on the PTZ reactivity contrasting with expectations. Therefore, even if the role of the acid sites is clearly established in the ionization process, we must also assume that the very low PTZ ionization potential value (I.P. = 6.73 eV) probably favours the ionization process in less polarizing environment. Indeed, PTZ ionization has been recently shown to take place in totally dealuminated silicalite-1.37 In contrast, when the ionization potential of the guest molecule is higher than 8 eV, spontaneous ionization requires a preliminary thermal treatment above 873 K under O2 to occur.31,32 In addition note that the presence of extraframework aluminium, which was detected in low amount in the zeolite samples investigated here, is sometimes put forward to account for the observation of spontaneous ionization. In this work as well as in some other studies,23 no significant change of spontaneous ionization was observed when increasing the Al extraframework content.
The charge separated states spontaneously created show very long lifetimes within H-FER, H-ZSM-5 and H-MOR. Electron trapping in a site lying far away from the PTZ•+ species prevents rapid direct charge recombination as observed in solution.29 Moreover, the diffusion of the radical cation within the channels is probably closely associated with the electron transport and these processes are significantly hindered by the long range electrostatic interactions through the resulting electric field, on one hand, and by the close match between the PTZ•+ size and inner porous volume, in the other hand.35,61 Additionally, the DRUVv spectra recorded as a function of time show clearly that the transient species do not evolve in the same way within H-FER, H-ZSM-5 and H-MOR. The spectral data provide evidence of the PTZ2+ dication formation for several months in each type of zeolite morphology but the PTZ2+ spectral concentration was found to increase dramatically on going from H-MOR to H-FER. Then, even if the kinetics of the ionization process is much faster in the larger pore H-MOR, the charge-separated state appears to be markedly less stabilized in this zeolite. In contrast, the higher confinement and consequently the higher polarizing effects offered by the 10-MR channels of H-FER induces the spontaneous second oxidation of PTZ into PTZ2+ with high yield. Within H-ZSM-5, the dication is also significantly produced but its spectral concentration remains slightly lower than that of PTZ•+. Thus, the relative proportion of PTZ2+ with respect to PTZ•+ depends undoubtedly on the pore opening dimension. The DRUVv absorption spectra obtained for the H-FER zeolite (Fig. 1) highlight the rapid emergence of the dication spectral feature at about 660 nm. The kinetic curves deduced from the chemometric analysis of the spectral data set obtained after mixing PTZ and H-FER (Fig. 2B) indicate that PTZ2+ becomes the major species after about 50 days. Therefore, if we assume a decrease of the distance between the charge compensating H+ cation and the PTZ species on going from H-MOR to H-FER and a parallel increase of the polarization energy, we expect higher second ionization efficiency in H-FER and H-ZSM-5 than in H-MOR. These results demonstrate the crucial role of both the confinement effect and the polarizing environment on the yield of PTZ intrazeolite second oxidation. However, note that the confinement effect within the internal void space of H-MOR is strong enough to induce quasi total PTZ first ionization.
The charge separated state created in these channel type zeolites exhibit exceptional stability for months and even years. However, the experiments show that after about 1.5 to 2 years for H-FER and H-ZSM-5 and less than 100 days for H-MOR, the relative spectral concentrations of PTZ•+ and PTZ2+ in the three zeolites undoubtedly decrease. Nevertheless, the cation recombination depends drastically on the zeolite morphology. The reaction is very slow in H-FER and H-ZSM-5 and PTZ•+ and PTZ2+ are still clearly observed after 2 years. In contrast, the larger pores of H-MOR increase the diffusion process and the probability of charge recombination. Therefore, it appears from the above mechanisms that PTZ•+ and PTZ2+ have very long lifetime but are only intermediates in the reaction and that the thermodynamically stable end product is the occluded PTZ molecule. Its formation kinetics depends on the coupled effects of both the inner space confinement and the H+ polarization energy. Indeed, the very slow charge recombination observed in acid zeolite after spontaneous ionization is highly correlated to the slow diffusion of the ionized species. After mixing, the spontaneous ionization occurs in parallel to the diffusion process. Then, diffusion of ionized species and subsequent electron transfer take place but the motions and charge transfer or recombination are markedly hampered by the very high intrazeolite electrostatic field. Consequently, the formation of the thermodynamically end product is drastically delayed and we assume that this behavior is mainly due to the extremely slow kinetics of the reaction and do not depend on a thermodynamic factor.
The diffusion processes of such charged species have to be compared with the behavior observed for neutral molecules when they penetrate into the host material. The diffusion of molecules depends mainly on the size of the guest molecule with respect to the pore diameter. Therefore, due to these steric constraints, diffusion takes place more or less rapidly before reaching equilibrium. Nevertheless, the diffusion of these uncharged species remains considerably faster than the diffusion of radicals which is drastically hindered by electrostatic interactions.
Moreover, it should be also interesting to compare this extremely slow kinetics observed after spontaneous ionization with the relatively faster reactions obtained after photoionization of polyaromatics within similar channel type zeolites. Photolysis induces the ionization of molecules located in the preferred sorption site and consequently no or very weak molecule diffusion is expected after ionization so that recombination only involves faster electron transfers.
Conclusion
Diffuse reflectance UV-visible and Raman experimental data show the PTZ sorption and spontaneous ionization in the straight channels of H-FER, H-ZSM-5 and H-MOR. The PTZ incorporation within the channels of these three zeolite topologies is also confirmed by molecular modeling. The very easy spontaneous ionization process has to be correlated with the low potential value of PTZ and occurs probably quasi simultaneously with the sorption. The experimental data demonstrate the crucial role of the combined effects of confinement and local electrostatic field on the sorption and charge separation kinetics. However, the DRUVv data show that the larger pores of H-MOR favour faster diffusion and ionization of PTZ. The mixing of PTZ with H-FER, H-ZSM-5 or H-MOR leads systematically to the PTZ•+ formation in quantity but the system advancement differs significantly as a function of channel structure. The second PTZ ionization was also observed within the three zeolites but the PTZ2+ spectral concentration appears to be much higher within the narrower pore of H-ZSM-5 and especially of H-FER. The higher proximity between the local electrostatic field and the radical cation is invoked to explain this feature.The exceptional stability of these very long lived charge separated state is then explained by the restricted mobility of the guest molecule in the narrow channels as well as the compartmentalization of the trapped electron away from the initial site of PTZ ionization. However, the slow but undoubted decrease of PTZ•+ and PTZ2+ spectral concentrations after more than one year in H-FER and H-ZSM-5 and only about 100 days in H-MOR demonstrates that these cations act only as intermediates. The occluded PTZ corresponds to the thermodynamically stable final product and the kinetics of formation is found to be very slow.
References
- S. Takagi, M. Eguchi, D. A. Tryk and H. Inoue, Langmuir, 2006, 22, 1406 CrossRef CAS
.
- J. K. Thomas, Chem. Rev., 2005, 105, 1683 CrossRef CAS
- S. L. Williams, I. Kirkpatrick and D. R. Worrall, Photochem. Photobiol. Sci., 2010, 9, 937 CAS
- S. L. Williams, D. R. Worrall, I. Kirkpatrick, A. Vancea and J. Pan, Photochem. Photobiol. Sci., 2011, 10, 84 CAS
- D. R. Worrall, S. L. Williams and T. Ganguly, Photochem. Photobiol. Sci., 2006, 5, 844 CAS
- D. R. Worrall, S. L. Williams, F. Wilkinson, J. E. Crossley, H. Bouas-Laurent and J. P. Desvergne, J. Phys. Chem. B, 1999, 103, 9255 CrossRef CAS
- X. Y. Zhu, Q. Yang and M. Muntwiler, Acc. Chem. Res., 2009, 42, 1779 CrossRef CAS
- Z. E. X. Dance, M. J. Ahrens, A. M. Vega, A. B. Ricks, D. W. McCamant, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2008, 130, 830 CrossRef CAS
- S. Hashimoto, J. Chem. Soc., Faraday Trans., 1997, 93, 4401 RSC
- K. B. Yoon, Chem. Rev., 1993, 93, 321 CrossRef CAS
- K. Yoon, Y. Park and J. Kochi, J. Am. Chem. Soc., 1996, 118, 12710 CrossRef CAS
- M. Alvaro, P. Atienzar, A. Corma, B. Ferrer, H. Garcia and M. T. Navarro, J. Phys. Chem. B, 2005, 109, 3696 CrossRef CAS
- M. A. Coutant, J. R. Sachleben and P. K. Dutta, J. Phys. Chem. B, 2003, 107, 11000 CrossRef CAS
- J. C. Scaiano and H. Garcia, Acc. Chem. Res., 1999, 32, 783 CrossRef CAS
- H. Zhang, C. S. Rajesh and P. K. Dutta, J. Phys. Chem. C, 2009, 113, 4623 CAS
- J. V. Caspar, V. Ramamurthy and D. R. Corbin, J. Am. Chem. Soc., 1991, 113, 600 CrossRef CAS
- H. Garcia and H. D. Roth, Chem. Rev., 2002, 102, 3947 CrossRef CAS
- V. Ramamurthy, J. V. Caspar and D. R. Corbin, J. Am. Chem. Soc., 1991, 113, 594 CrossRef CAS
- G. Calzaferri, S. Huber, H. Maas and C. Minkowski, Angew. Chem., Int. Ed., 2003, 42, 3732 CrossRef CAS
- A. Moissette, S. Marquis, D. Cornu, H. Vezin and C. Brémard, J. Am. Chem. Soc., 2005, 127, 15417 CrossRef CAS
- H. Ikeda, T. Nomura, K. Akiyama, M. Oshima, H. D. Roth, S. Tero-Kubota and T. Miyashi, J. Am. Chem. Soc., 2005, 127, 14497 CrossRef CAS
- X. Liu, K. K. Iu, J. K. Thomas, H. He and J. Klinowski, J. Am. Chem. Soc., 1994, 116, 11811 CrossRef CAS
- S. Marquis, A. Moissette, H. Vezin and C. Brémard, C. R. Chim., 2005, 8, 419 CrossRef CAS
- A. Bhan and E. Iglesia, Acc. Chem. Res., 2008, 41, 559 CrossRef CAS
- A. Moissette, S. Marquis, I. Gener and C. Brémard, Phys. Chem. Chem. Phys., 2002, 4, 5690 RSC
- S. Marquis, A. Moissette, H. Vezin and C. Bremard, J. Phys. Chem. B, 2005, 109, 3723 CrossRef CAS
- S. Marquis, A. Moissette, M. Hureau, H. Vezin and C. Brémard, J. Phys. Chem. C, 2007, 111, 17346 CAS
- H. Vezin, A. Moissette and C. Brémard, Angew. Chem., Int. Ed., 2003, 42, 5587 CrossRef CAS
- A. E. Keirstead, N. P. Schepp and F. L. Cozens, J. Phys. Chem. C, 2007, 111, 14247 CAS
- D. W. Werst, P. Han and A. D. Trifunac, Radiat. Phys. Chem., 1998, 51, 255 CrossRef CAS
- A. F. Bedilo and A. M. Volodin, Kinet. Catal., 2009, 50, 314 CrossRef CAS
- T. M. Leu and E. Roduner, J. Catal., 2004, 228, 397 CrossRef CAS
- M. J. Nash, A. M. Shough, D. W. Fickel, D. J. Doren and R. F. Lobo, J. Am. Chem. Soc., 2008, 130, 2460 CrossRef CAS
- A. Moissette, H. Vezin, I. Gener and C. Bremard, J. Phys. Chem. B, 2003, 107, 8935 CrossRef CAS
- M. Hureau, A. Moissette, S. Marquis, C. Brémard and H. Vezin, Phys. Chem. Chem. Phys., 2009, 11, 6299 RSC
- A. Moissette, R. F. Lobo, H. Vezin, K. A. Al-Majnouni and C. Bremard, J. Phys. Chem. C, 2010, 114, 10280 CAS
- A. Moissette, F. Luchez, C. Brémard, H. Vezin and M. Hureau, Phys. Chem. Chem. Phys., 2009, 11, 4286 RSC
- J. Daub, R. Engl, J. Kurzawa, S. E. Miller, S. Schneider, A. Stockmann and M. R. Wasielewski, J. Phys. Chem. A, 2001, 105, 5655 CrossRef CAS
- A. Knorr and J. Daub, Angew. Chem., Int. Ed. Engl., 1997, 36, 2817 CrossRef CAS
- C. M. Gooley, H. Keyzer and F. Setchell, Nature, 1969, 223, 80 CrossRef CAS
- M. Bevilacqua, T. Montanari, E. Finocchio and G. Busca, Catal. Today, 2006, 116, 132 CrossRef CAS
- A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS
- R. Gounder and E. Iglesia, J. Am. Chem. Soc., 2009, 131, 1958 CrossRef CAS
- W. Windig, Chemom. Intell. Lab. Syst., 1997, 36, 3 CrossRef CAS
- W. Windig, B. Antalek, J. L. Lippert, Y. Batonneau and C. Bremard, Anal. Chem., 2002, 74, 1371 CrossRef CAS
- R. Tauler, Chemom. Intell. Lab. Syst., 1995, 30, 133 CrossRef CAS
- J. A. VanAllan, G. A. Reynolds and R. E. Adel, J. Org. Chem., 1962, 27, 1659 CrossRef CAS
- O. Brede, A. Maroz, R. Hermann and S. Naumov, J. Phys. Chem. A, 2005, 109, 8081 CrossRef CAS
- D. M. Chapman, A. C. Buchanan III, G. P. Smith and G. Mamantov, J. Am. Chem. Soc., 1986, 108, 654–663 CrossRef CAS
- H. J. Shine and E. E. Mach, J. Org. Chem., 1965, 30, 2130 CrossRef CAS
- E. Wagner, S. Filipek and M. K. Kalinowski, Monatsh. Chem., 1988, 119, 929 CrossRef CAS
- H. Jian, J. Xiang, K. Sun, J. Sun, C. Chen, B. Zhou, Y. Liu and G. Xu, J. Colloid Interface Sci., 2000, 229, 212 CrossRef CAS
- R. E. Hester and K. P. J. Williams, J. Chem. Soc., Perkin Trans. 2, 1981, 852 RSC
- D. Pan and D. L. Phillips, J. Phys. Chem. A, 1999, 103, 4737 CrossRef CAS
- G. Sarata, Y. Noda, M. Sakai and H. Takahashi, J. Mol. Struct., 1997, 413, 49 CrossRef
- G. Sarata, M. Sakai and H. Takahashi, J. Raman Spectrosc., 2000, 31, 785 CrossRef CAS
- S. Hashimoto, J. Photochem. Photobiol., C, 2003, 4, 19 CrossRef CAS
- F. Belhadj, A. Moissette, C. Brémard and M. Hureau, ChemPhysChem, 2011, 12, 1378 CrossRef CAS
- L. Domokos, L. Lefferts, K. Seshan and J. A. Lercher, J. Mol. Catal. A: Chem., 2000, 162, 147 CrossRef CAS
- A. Bhan, A. D. Allian, G. J. Sunley, D. J. Law and E. Iglesia, J. Am. Chem. Soc., 2007, 129, 4919 CrossRef CAS
-
J. Krager
D. M. Ruthven, ‘Diffusion in Zeolites and other Microporous Solids’, Wiley, New York, 1992 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00220a |
| This journal is © The Royal Society of Chemistry 2011 |