Facile synthesis of 5-(alkylidene)thiophen-2(5H)-ones. A new class of antimicrobial agents†
Tore
Benneche
*a,
Gunnar
Herstad
a,
Marianne
Rosenberg
a,
Synnøve
Assev
b and
Anne Aamdal
Scheie
b
aDepartment of Chemistry, University of Oslo, PO. Box 1033, Blindern, 0315 Oslo, Norway. E-mail: toreben@kjemi.uio.no
bDepartment of Oral Biology, Faculty of Dentistry, University of Oslo, Norway
First published on 8th August 2011
Abstract
5-(Chloromethylene)- and 5-(bromoalkylidene)thiophen-2(5H)-ones are easily prepared from readily available 2-acyl-5-methoxythiophenes in one step by reaction with excess acetyl chloride or acetyl bromide. Other 5-(methylidene)thiophen-2(5H)-ones have been made from 5-(bromomethylene)thiophene-2(5H)-one by treatment with different nucleophiles. Eighteen thiophenones were tested for capacity to reduce biofilm formation by the marine bacterium V. harveyi. All but three compounds reduced biofilm formation markedly. For all effective compounds but one, the relative biofilm reducing capacity was greater than inhibition of bacterial planktonic growth.
Introduction
Microorganisms prefer a life in biofilm. A biofilm is an aggregate of microorganisms in which cells adhere to each other and to a surface, rendering them more resistant to antimicrobial agents. The ability to form biofilm is widely spread among bacteria. The presence of a biofilm may have substantial implications in fields ranging from various industrial processes to health related fields like medicine and dentistry.1 In many bacteria, the mechanisms of biofilm formation include a cell-density regulated gene expression called quorum sensing (QS).2 QS communication is induced by signal molecules that bacteria produce and sense leading to regulation of a variety of physiologic functions. The marine bacterium Vibrio harveyi, which is a pathogen in aquaculture, regulate biofilm formation and toxin production via QS.3 QS represents an interesting novel target for control of biofilm related problems, and has lately created great interest in the scientific community as a method predicted not to lead to development of bacterial resistance.4A number of (Z)-5-(bromomethylidene)furan-2(5H)-ones (1, Fig. 1) are known to interfere with bacterial QS and to inhibit biofilm formation.3 There is some information about structure–activity relationship (SAR) concerning side chains in furan-2(5H)-ones,5 but very little is known about SAR in relation to the ring system. It is, however, known that nitrogen analogues (pyrrolones, 2, Fig. 1) of 5-(bromomethylene)furan-2(5H)-ones are active in interfering with bacterial QS.6 In order to investigate other systems, we decided to synthesize sulphur analogues of 1 (thiophenones, 3, Fig. 1).
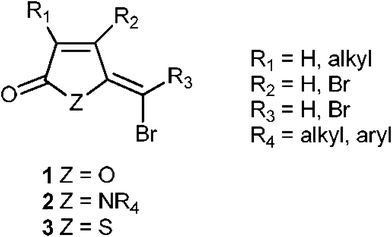 | ||
| Fig. 1 Furanones, pyrrolones and thiophenones that may inhibit quorum sensing. | ||
5-Alkylidenethiophen-2(5H)-ones have been prepared by elimination from 5-(1-hydroxyalkyl)-2-alkoxythiophenes under acidic conditions7 (A, Scheme 1), by condensation of 5-unsubstituted thiophen-2(5H)-ones with aldehydes under basic conditions8 (B) and by hydride abstraction from 2-t-butoxy-5-(cyclohepta-2,4,6-trienyl)thiophene9 (C).
 | ||
| Scheme 1 | ||
A number of the naturally occurring furanones which are QS inhibitors, have a 5-bromomethylene substituent.4c,10 The importance of this substituent has also been demonstrated in synthetic furanones.5 Hence we first directed our efforts towards the synthesis of 5-(bromomethylene)thiophen-2(5H)-ones. In addition we wanted to see if other 5-alkylidene analogues of 3 had a similar biological profile as the 5-bromomethylene compounds. We were particularly interested in 5-(alkylidene)thiophen-2(5H)-ones having a heteroatom substituent in the alkylidene group because the resulting thiophenones would be similar to known QS inhibitors from the furanone series.11 None of the existing methods mentioned above (A–C, Scheme 1)7–9 seemed to be suitable for the synthesis of such 5-(alkylidene)thiophen-2(5H)-ones.
Results and discussion
Chemistry
We noted that the readily available 2-formyl-5-methoxythiophene12 (4a, Scheme 2) had all the carbon atoms in place for the synthesis of 5-(halomethylene)thiophen-2(5H)-one (3/6, Table 1). All that was needed was a transformation of the methoxy group into an oxo group and the formyl group into a halomethylene group. Previous synthetic efforts have shown that a methoxy group in 2-position of a thiophene ring can be transformed into an oxo group by protonating a substituent in the 5-position with aqueous acid.7b,7c Thus we needed to increase the electron withdrawing properties of the formyl group in order to activate for the transformation of the methoxy substituent into an oxo group.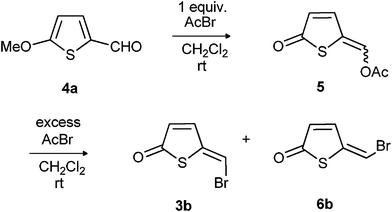 | ||
| Scheme 2 | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Compound 3/6 | R | X | Conditions | Yield 3/6 (%) | Z/E |
| a Partly substitution in the R group, see Experimental; b the product was isolated as the bromide; c only Z-isomer isolated; d a mixture of E- and Z-isomers; e from 1H NMR of the crude product; f 28% of (Z)-(4-(tert-butyl)-5-oxothiophen-2(5H)-ylidene)methyl acetate was isolated. | ||||||
| 1 | a | H | Cl | CDCl3, rt, 3d | 49 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | b | H | Br | CH2Cl2, rt, 4d | 71 | 5:1 |
| 3 | b | H | Br | CDCl3, 60 °C, 3h | 61 | 11:1 |
| 4 | c | CH2Cl | Cl | CDCl3, 60 °C, 24h | 49 | 15:1 |
| 5 | d | CH2Cl a | Br | CH2Cl2, rt, 3d | 49 | — |
| 6 | e | CH3CHCl/Brb | Br | CHCl3, rt, 4d | 39c | — |
| 7 | f | Et | Br | CH2Cl2, rt, 8d | 41d | — |
| 8 | g | i-Pr | Br | CDCl3, rt, 7d | 41c | 6:1e |
| 9 | h | t-Bu | Br | CDCl3, 60 °C, 8h | 88 | 7:1 |
| 10 | h | t-Bu | Br | CDCl3, rt, 2d | 29f | 9:1 |
| 11 | i | Br | Br | CDCl3, 60 °C, 3h | 72 | 8:1 |
| 12 | j | 2-thienyl | Br | CH2Cl2, rt, 3d | 32c | 9:1e |

First we tried boron tribromide since this reagent is known to cleave aromatic methoxy groups and to transform aromatic aldehydes into dibromides.13 A complex reaction mixture was, however, obtained and we could not isolate any 5-(bromomethylene)thiophen-2(5H)-one from this mixture. Next we tried dry hydrogen bromide in dichloromethane, which gave (Z)- and (E)-5-(bromomethylene)thiophen-2(5H)-one (3b/6b, Scheme 2) in a yield of 10% and 2%, respectively. Better results were obtained with excess acetyl bromide in dichloromethane, which gave a mixture of (Z)- and (E)-5-(bromomethylene)thiophen-2(5H)-one in 71% yield (Table 1, entry 2). Phosphorus tribromide can also be used but the reaction is not clean and the yields are lower than with acetyl bromide.
By following the reaction (TLC, 1H NMR) we observed that the enol acetate 5 was formed before the 5-bromomethylene compounds 3b/6b (Scheme 2). Isolated enol acetate 5 could be transformed into compounds 3b and 6b by treatment with excess of acetyl bromide. The formation of the enol acetate 5 is fairly rapid compared to the consecutive formation of the bromo compound.
It is not necessary to isolate the enol acetate 5 in this reaction, the 5-bromomethylene thiophenones 3b and 6b could be formed in one pot. The reaction could be done by heating at 60 °C for 3h with a large excess of acetyl bromide (Table 1, entry 3), but the yield is better if the aldehyde 4a is treated with two equivalents acetyl bromide for 24 h before a large excess of acetyl bromide is added and the mixture stirred at room temperature for 4 days (Table 1, entry 2).
A number of derivatives of 2-formyl-5-methoxythiophene 4a were made in order to test their reaction with acyl halides (Scheme 3).
 | ||
| Scheme 3 | ||
The 4-position in compound 4a is activated towards electrophilic attack. Thus compounds 4b, c (R = iPr, tBu) were made by Friedel–Crafts alkylation (Scheme 3). Chloromethylation of 4a by chloromethyl ethyl ether in the presence of titanium tetrachloride gave the compounds 4f (R = H). All attempts to make 4g (R = Me) by the same method failed. This compound was, however, made by a multistep procedure: bromination of the aldehyde 4a with NBS gave compound 4d; a Stille reaction with tributylvinylstannane gave the alkene 4e which could be reacted with dry hydrogen chloride to the compound 4g or reduced with hydrogen to the compound 4i. The bithiophenyl compound 4h was made by another Stille reaction. The imine 7 and the alkene 8 were synthesized from the aldehyde 4a by standard methods.
The formation of the thiophenones 3b/6b (Scheme 2), which do not have a substituent in the 3 position, seems more sensitive to the reaction conditions than the formation of the 3-substituted thiophenones (Table 1, entries 4–12). Thus treatment of the aldehyde 4a with acetyl bromide gives immediately a black solution while reaction of the aldehyde 4c gives a clear yellow solution even after heating at 60 °C for 8 h (entry 9). The yield in this case is also better than the corresponding room temperature reaction (entry 10). The reason for this might be that the thioester functionality is protected to some degree by a substituent in the 3-position. So heating may give better yields in this reaction if the starting material does not decompose at an elevated temperature.
A solution of the pure E-isomer 6b (Scheme 2) will isomerise into a mixture of E/Z-isomers with the Z-isomer as the major product. This indicates that the Z-isomers 3 are thermodynamically more stable than the E-isomers 6.This isomerisation is fairly rapid at room temperature and can in some cases make it difficult to isolate the pure E- and Z-isomer.
By using acetyl chloride instead of acetyl bromide the corresponding chloride could be obtained (Table 1, entries 1 and 4). Thionyl chloride can also be used, but the yields are inferior to acetyl chloride.
A number of 4-substituted 2-formyl-5-methoxythiophenes (4) could be transformed into 5-(halomethylene)thiophen-2(5H)-ones (3 and 6) by the method described above (Table 1).
We were interested in having an alcohol function in the side chain of the thiophenone for several reasons: (1) to try to improve the solubility in water; (2) to test the activity of the hydroxy compoundper se and (3) to use the hydroxy group as an anchorage point for a linker to a polymer or a metal surface. Generally the thiophenones have very little stability under aqueous basic conditions—probably due to the opening of the ring system (e.g.sodium methoxide in methanol cleaves the thioester). The compounds have much greater stability under neutral or acidic conditions. Simple hydrolysis of the compounds 3d, e under aqueous acidic conditions, however, gave a complex mixture of starting material, wanted product and other unidentified compounds. A much cleaner reaction was obtained if the compounds were treated with silver triflate in the presence of water (Scheme 4). The compounds 9a and 9b were isolated in 69% and 72% yield, respectively.
 | ||
| Scheme 4 | ||
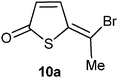
We thought that a natural extension of having a 2-formyl group in the thiophene ring, would be to try other acyl groups in this position. Hence 2-acetyl-5-methoxythiophene was prepared according to the literature14 and treated with acetyl bromide. E- and Z-5-bromoethyliedenethiophen-2(5H)-one 10a and 11a were formed in 64% yield and with the E-isomer as the main product (Table 2, entry 1). This is opposite of what is obtained in the 2-formyl series, where the Z-isomer is usually the main product. With 2-benzoyl-5-methoxythiophene15 the Z-isomer (10c) was again the main product (entry 3). In this case also 14% of the corresponding 5-enol acetate was isolated, indicating that this reaction goes through an enol acetate as an intermediate. Having a trifluoroacetyl group in the 2-position resulted in an unreactive thiophene derivative (entry 2). Thus 2-trifluoroacetyl-5-methoxythiophene16 remained unchanged after reaction with a large excess of acetyl bromide in refluxing CDCl3 for 24 h. This indicates that in order for this reaction to occur it must be possible to acetylate the acyl oxygen, which in this case may be difficult because of the electron withdrawing property of the trifluoromethyl group. The acetylation of the acyl oxygen is only the first step in the formation of a bromoalkylidene compound. The second step is the exchange reaction of the acetoxy group for a bromide and this may be a slower reaction than the acetylation. This is seen in the reaction of 2-formyl-5-methoxyfurane17 with acetyl bromide where the corresponding enol acetate is readily formed, but the enol acetate does not react with acetyl bromide to give the corresponding bromide (Scheme 5).19
 | ||
| Scheme 5 | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Compound 10/11 | R | X | Y | Yield % 10/11 | Z/E-ratio |
| a Stereochemistry based on 1H NMR chemical shifts, coupling constants and Rf value of the isomers; b 14% of the corresponding Z-enol acetate (10 Y = OAc) was isolated; c Z-isomer as indicated by NOESY experiment; d caused by HBr in the acetyl bromide. | ||||||
| 1 | a | CH3 | O | Br | 64 | 0.4:1a |
| 2 | b | CF3 | O | Br | 0 | — |
| 3 | c | Ph | O | Br | 45b | 2.2:1a |
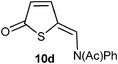
| 4 | d | H | NPh | N(Ac)Ph | 60c | — |
| 5 | e | H | CBr2 | CHBr2d | 47c | — |
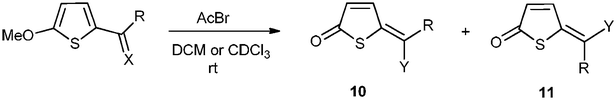
We were also interested to see whether 5-methoxythiophenes having 2-substituents with double bonds other than C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O could react in this reaction. First we prepared the imine 7 (Scheme 3) which reacted with a slight excess of acetyl bromide to give the amide 10d in 60% yield (Table 2, entry 4). Only one isomer (Z) was observed.
O could react in this reaction. First we prepared the imine 7 (Scheme 3) which reacted with a slight excess of acetyl bromide to give the amide 10d in 60% yield (Table 2, entry 4). Only one isomer (Z) was observed.
Attempt to acetylate the double bond in the Wittig product 8 (Scheme 3) with acetyl bromide in a Friedel–Crafts reaction without any catalyst, gave only the addition product 10e (entry 5). The acetyl bromide solution was not freshly distilled before use, so it probably contained hydrogen bromide which accounts for the product formed.
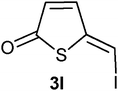
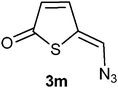
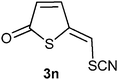
A probable mechanism for the transformation of compound 5 into compounds 3b and 6b, is a Michael type addition of bromide to the exocylic double followed by elimination of the acetate group. This implies that it should be possible to exchange the bromo substituent in compound 3 and 6 with other nucleophiles. Indeed, treatment of 3b with one equivalent of tetrabutylammonium chloride in deuterated chloroform resulted in a complete exchange of the bromo substituent for a chloro group after 5 h at room temperature (Table 3, entry 1). This reactivity gave us a way to prepare 5-(iodomethylene)thiophen-2(5H)-one (3l), 5-(azidomethylene)thiophen-2(5H)-one (3m) and 5-(thiocyanatomethylene)thiophen-2(5H)-one (3n) (entries 3–5). In addition the 3-bromosubstituted thiophene-2(5H)-ones 3k and 3o were made (entries 2 and 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | X-Y | R | Conditions | Yield%a |
| a Small amounts of the E-isomer were observed; b according to 1H NMR. | |||||
| 1 | a | n-Bu4N-Cl | H | CDCl3, rt, 5h | Quant.b |
| 2 | k | n-Bu4N-Cl | Br | CDCl3, rt, 24h | 86 |
| 3 | l | Na-I | H | Acetone, rt, 3d | 73 |
| 4 | m | Na-N3 | H | DMF, 0 °C, 20 min | 89 |
| 5 | n | NH4-SCN | H | Acetone, rt, 4h | 62 |
| 6 | o | NH4-SCN | Br | Acetone, rt, 30 min | 78 |

The stereochemistry assignment of the exocyclic double bond in 3 and 6 is based on coupling constants and chemical shift in 1H NMR, on NOESY experiments and on Rf-values. For example, the E-isomers 6a and 6b show a five-bond coupling constant between H3 and H1′ of about 1.7 Hz and a four-bond coupling between H4 and H1′ of about 0.8 Hz. This is in accordance with the corresponding coupling constants for cis-1-methoxy-1,3-butadien.18 The Z-isomers 3a and 3b show a much lower long range coupling constant (JH3/H1′ = 0.6 Hz). This is also in accordance with the corresponding coupling constants for trans-1-methoxy-1,3-butadiene.17 In all cases, so far observed, the chemical shift value for the H4-proton in the E-isomer has a higher δ-value than the δ-value for H4-proton in the Z-isomer. The difference between the E- and Z-isomer range from δ 0.22 to 0.62 (Table 4)
|
|
|||||
|---|---|---|---|---|---|
| X | R | R1 | H4-E (δ) | H4-Z (δ) | H4-E - H4-Z (δ) |
| a Ref. 19. | |||||
| OAc | H | H | 7.99 | 7.61 | 0.38a |
| Cl (3a/6a) | H | H | 8.02 | 7.52 | 0.50 |
| Br (3b/6b) | H | H | 7.95 | 7.48 | 0.47 |
| I (3l/6l) | H | H | 7.78 | 7.44 | 0.34 |
| Br (10a/11a) | H | Me | 7.89 | 7.67 | 0.22 |
| Br (10c/11c) | H | Ph | 8.12 | 7.49 | 0.62 |
| Cl (3c/6c) | CH2Cl | H | 8.02 | 7.55 | 0.47 |
| Br (3h/6h) | t-Bu | H | 7.56 | 7.13 | 0.43 |
| Br (3i/6i) | Br | H | 8.09 | 7.65 | 0.44 |

Also in all cases so far observed the Rf-value of E-isomer is higher than the Rf-value of the Z-isomer when the chromatography is done on silica plates using a mixture of hexane and ethyl acetate for elution.
Biofilm and planktonic growth inhibition
The various compound concentrations inhibiting 50% of biofilm formation (BIC50) and of planktonic growth (PIC50) in Vibrio harveyiBB120 are summarized in Table 5. Only the Z-isomers (3 of the E/Z-isomers 3 and 6 have been tested. The reason for this is that the E-isomers 6 may partly isomerize to the more stable Z-isomers 3 (vide supra) during the test. We also have indication that the Z-isomers are more active than the E-isomers, e.g.3a has a higher degree of inhibition of biofilm formation by S. epidermidis than 6a (data not shown).












Compounds 5, 9a, and 10d had neither effect on biofilm formation nor planktonic growth at the concentrations tested (5–20 μM, 5–10 μM and 100 μM respectively). Apart from compounds 5, 9a and 10d, all compounds had inhibitory effects on both biofilm formation and planktonic growth. The inhibitory effect was generally more marked for biofilm formation than for planktonic growth, except for compound 3l (Table 5). At 37 μM concentration or less, compounds 3b, f, g, l, m, n, o, 10a and 11c inhibited biofilm formation by 50%. At 50 μM and 61 μM, 3j and 3k also inhibited biofilm formation by 50%. The compounds 3b, f, l, n, o and 11c were highly effective, inhibiting biofilm formation more than 50% at 20 μM or less (Table 5). Compound 3l seemed to be the most efficacious, reducing biofilm formation by 67% already at 5 μM. The biofilm inhibitory effect of this compound was probably caused by a growth inhibitory effect since the effect on planktonic cells was high. For practical purposes, the choice of compound would depend on field of and mode of application, compound efficacy, toxicity and stability. In view of risk of bacterial resistance development, a compound that inhibits biofilm formation without killing the bacteria would probably be desirable, since it would exert no or low selective pressure.
(Z)-5-(Bromomethylene)thiophen-2(5H)-one (3b) was more effective in inhibiting biofilm formation than the corresponding furanone (F202) (Table 5). This is in line with previous data comparing the inhibitory effect of (Z)-5-(bromomethylene)furan-2(5H)-one (F202)- and (Z)-5-(bromomethylene)thiophen-2(5H)-one (3b)-coated surfaces on biofilm formation by Staphylococcus epidermidis.20 This latter study also give evidence for the assumption that the thiophenones inhibit quorum sensing.
Conclusion
We have shown that 5-(chloromethylidene)- and 5-(bromoalkylidene)thiophen-2(5H)-ones can easily be synthesized in one step from readily available 2-acyl-5-methoxythiophenes. Further that a range of 5-(alkylidene)thiophen-2(5H)-ones can be prepared from 5-(bromomethylidene)thiophen-2(5H)-one by reaction with different nucleophiles. While the biofilm- and planktonic inhibitory capacity of the selected thiophenones have been documented, the mechanism of action, stability in various environments and possible eco- and human toxicity remains to be elucidated. The thiophenones represent a promising, novel class of anti-biofilm/anti-bacterial agents.Experimental
Biofilm and planktonic growth inhibition assay
Biofilm inhibition was assessed as reduction in biofilm mass produced by the marine bacterium Vibrio harveyiBB120 (a kind gift from Prof. BL. Bassler). Vibrio harveyiBB120 was kept as a frozen stock at −70 °C. Before use, Vibrio harveyiBB120 was reinoculated and incubated for 8h in fresh synthetic seawater growth medium (10 g L−1 beef extract, 10 g L−1peptone, 30 g L−1 sea salt (Sigma-Aldrich, MO, USA) in a shaking water bath at 30 °C. The culture was then reinoculated into fresh medium and incubated over night. The culture was diluted 1:100 in fresh medium. Thiophenones were added to final concentrations varying from 5 μM to 300 μM concentrations. 200 μl incubation mixtures were incubated in 96 well polystyrene microtiter plates (Nunc, Roskilde, Denmark) with pegged lids (Nunc-TSP, Nunc, Roskilde, Denmark) according to the Calgary Biofilm Device method.21 After 4 h incubation, unattached cells were removed by rinsing the pegged lids twice in 0.9% saline. The biofilm on the pegs was then stained for 10 min with a 0.1% solution of safranin and washed with 0.9% saline until stain-free washings were obtained. Bound stain was released using 30% glacial acetic acid, and optical density (OD) at 530 nm was measured in a Synergy™ HT multi-detection microplate reader (Bio-Tek Instruments Inc. Vermont, USA). Biofilm reduction was calculated as % compared to control.
Inhibition of planktonic (free floating bacteria) growth was assayed in parallel by growing Vibrio harveyiBB120 cultures with thiophenone in low-cell binding plates (Nalge Nunc Int. Tokyo, Japan) and measuring optical cell density after 4 h at 600 nm. Planktonic growth inhibition was calculated as % reduction compared to control. The compound concentrations giving 50% reduction of biofilm formation and planktonic growth inhibition were calculated from dose response curves. As a control the biofilm and planktonic inhibitory capacities of (Z)-5-(bromomethylene)furan-2(5H)-one and (Z)-5-(bromomethylene)thiophen-2(5H)-one were compared using the same assays as described.
Chemistry
The 1H NMR and the 13C NMR spectra were recorded on Bruker Avance DPX instruments. Mass spectra, under electron impact conditions, were recorded at 70 eV ionizing energy on a Fision ProSpec instrument.(Z)-5-(Chloromethylene)thiophen-2(5H)-one 3a. Eluent Hexanes–EtOAc 5
:1; δH (200 MHz, CDCl3) 6.39 (1H, d, J 6.0, H1′) 6.85 (1H, s, H3) , 7.52 (1H, d, J 6.0, H4); δC (75 MHz; CDCl3) 123.3, 131.8, 142.3, 147.0, 193.3; m/z (EI) 148 (M++2, 37%), 146 (M+, 100), 120 (12), 118 (34), 111 (29), 83 (24); HRMS (EI). Calculated for C5H3ClOS 145.9593. Found 145.9597.
(E)-5-(Chloromethylene)thiophen-2(5H)-one 6a. Eluent Hexanes–EtOAc 5
:1; δH (200 MHz, CDCl3) 6.49 (1H, dd, 1H, J 6.4, 1.6, H3), 6.51 (1H, s, H1́), 8.02 (1H, dd, J 6.4, 1.6, H4![[) with combining cedilla]](https://www.rsc.org/images/entities/char_0029_0327.gif) δC (75 MHz; CDCl3) 119.2, 137.2, 138.6, 143.2, 194.3.
δC (75 MHz; CDCl3) 119.2, 137.2, 138.6, 143.2, 194.3.
(Z)-5-(Bromomethylene)thiophen-2(5H)-one 3b. Eluent Hexanes–EtOAc 5
:1; mp 67–71 °C; δH (200 MHz, CDCl3) 6.48 (1H, d, J 6.0, H1′) 7.10 (1H, s, H3) , 7.48 (1H, d, J 6.0, H4); δC (75 MHz; CDCl3) 112.7, 132.1, 145.1, 147.0, 193.0; m/z (EI) 192 (M++2, 100%), 190 (M+, 98), 164 (28), 162 (27), 111 (44), 83 (62); HRMS (EI). Calculated for C5H3BrOS 189.9088 Found 189.9087.
(E)-5-(Bromomethylene)thiophen-2(5H)-one 6b. Eluent Hexanes–EtOAc 5
:1; δH (200 MHz, CDCl3) 6.53 (1H, dd, 1H, J 6.0, 1.6, H3), 6.70–6.72 (1H, m, H1′), 7.95 (1H, dd, J 6.0, 1.6, H4); m/z (EI) 192 (M++2, 100%), 190 (M+, 96), 164 (30) 162 (28), 111 (37), 83 (41).
(Z)-3-(Chloromethyl)-5-(chloromethylene)thiophen-2(5H)-one 3c. Eluent Hexanes–EtOAc 5
:1; δH (200 MHz; CDCl3) 4.30 (2H, s, CH2Cl), 6.90 (1H, s, H1′), 7.55 (1H, s, H4); δC (75 MHz; CDCl3) 37.1, 124.1, 139.4, 141.6, 144.2, 191.1; m/z (EI) 198 (M++4, 14%), 196 (M++2, 69), 194 (M+ -100), 161 (36), 159 (94), 133 (23), 131 (57), 99 (50); HRMS (EI). Calculated for C6H4Cl2OS 193.9360. Found 193.9362.
(E)-3-(Chloromethyl)-5-(chloromethylene)thiophen-2(5H)-one 6c. Eluent Hexanes–EtOAc 5
:1; δH (200 MHz; CDCl3) 4.35 (2H, s, CH2Cl), 6.54 (1H, s, H1′), 8.02 (1H, s, H4); δC (75 MHz; CDCl3) 37.1 120.3, 135.6, 140.7, 143.0, 192.0; m/z (EI) 198 (M++4, 13%), 196 (M++2, 70), 194 (M+, 100), 161 (33), 159 (90), 133 (22) 131 (57), 99 (50); HRMS (EI). Calculated for C6H4Cl2OS 193.9360. Found 193.9359.
(Z)-5-(Bromomethylene)-3-(chloromethyl)thiophen-2(5H)-one (3d1) and (Z)- 3-(bromomethyl)-5-(bromomethylene)thiophen-2(5H)-one (3d2). A mixture of the title compound was obtained: 69% (Z)-5-(bromomethylene)-3-(chloromethyl)thiophen-2(5H)-one and 31%(Z)-3-(bromomethyl)-5- (bromomethylene)thiophen-2(5H)-one. Gradient elution 0–15% EtOAc in hexanes.
(Z)-5-(Bromomethylene)-3-(chloromethyl)thiophen-2(5H)-one 3d1. Mp 64–66 °C; δH (200 MHz; CDCl3) 4.26 (2H, s, CH2Cl), 7.15 (1H, s, H3), 7.51(1H, s, H4); δC (75 MHz; CDCl3) 36.7, 113.6, 141.7, 142.1, 144.3, 190.7; m/z (EI) 242 (M++4, 27%), 240 (M++2, 100), 238 (M+, 75), 205 (84), 203 (81), 177 (23), 175 (22), 145 (33), 143 (35); HRMS (EI). Calculated for C6H4BrClOS 237.8855. Found 237.8848.
(Z)-3-(Bromomethyl)-5-(bromomethylene)thiophen-2(5H)-one 3d2. Mp 53–56 °C; δH (200 MHz; CDCl3) 4.14 (2H, s, CH2Br), 7.15 (1H, s, H3), 7.51(1H, s, H4); δC (75 MHz; CDCl3) 21.5, 113.6, 141.8, 142.1, 144.6, 190.5; m/z (EI): 286 (M++4, 16%), 284 (M++2, 31), 282 (M+, 15), 205 (100), 203 (97), 145 (37), 143 (39), 96 (17); HRMS (EI). Calculated for C6H4Br2OS 281.8350. Found 281.8345.
(Z)-3-(1-Choroethyl)-5-(bromomethylene)thiophen-2(5H)-one 3e. Gradient elution 5–20% EtOAc in hexanes; δH (300 MHz; CDCl3) 1.92 (1H, d, J 7.0, CHCH3), 4.92 (1H, q, J 7.0, CHCH3), 7.14 (1H, s, H'), 7.50 (1H, s, H4); δC (75 MHz; CDCl3) 24.7, 37.4, 113.3, 142.1, 142.4, 147.2, 190.1; m/z (EI) 300(M++4, 7%), 298(M++2, 14), 296 (M+, 7), 219(100), 217 (99), 173 (8), 78 (32), 51 (10); HRMS (EI): Calculated for C7H6OSBr2 295.8506. Found 295.8505.
(Z)-3-(Ethyl)-5-(bromomethylene)thiophen-2(5H)-one 3f. Gradient elution 2.5–10% EtOAc in hexanes; δH (300 MHz; CDCl3) 1.15 (3H, t, J 7.5 , CH3), 2.33 (2H, tt, J 3.6, 7.4, CH2), 6.93 (1H, s, H'), 7.14 (1H, s, H4); δC (75 MHz; CDCl3) 12.1, 20.0 , 109.6, 140.8, 143.0, 148.4, 193.2; m/z (EI) 220 (M++2, 100%), 218 (M+, 96), 177 (27), 175 (26), 139 (30), 111 (51), 79 (74); HRMS (EI). Calculated for C7H7OSBr 217.9401. Found 217.9399.
(Z)-5-(Bromomethylene)-3-isopropylthiophen-2(5H)-one 3g. Eluent Hexanes-EtOAc 10
:1; δH (200 MHz; CDCl3) 1.16 [6H, d J 7.0, (CH3)2CH], 2.6–2.8 [1H, m, (CH3)2CH], 6.93 (1H, s, H1́), 7.11 (1H, s, H4); δC (75 MHz; CDCl3) 21.8, 27.1, 110.0, 140.2, 143.4, 153.3; m/z (EI) 234 (M++2, 100%), 232 (M+, 97%), 219 (45), 217 (44), 206 (15) 204 (15), 191 (24), 189 (22), 153 (94), 93 (99), 91 (35); HRMS (EI). Calculated for C8H9BrOS 231.9557. Found 231.9556.
(Z)-5-(Bromomethylene)-3-tert-butylthiophen-2(5H)-one 3h. Eluent Hexanes-EtOAc 10
:1; δH (200 MHz; CDCl3) 1.25 (9H, s, t-Bu), 6.90 (1H, s, H3), 7.13 (1H, s, H4); δC (75 MHz; CDCl3) 29.0, 34.3, 109.8, 140.9, 142.8, 154.6, 192.6; m/z (EI) 248 (M++2, 100%), 246 (M+, 98) 233 (57), 231 (56), 206 (54), 205 (49), 204 (53), 203 (41), 167 (83), 107 (92), 91 (69); HRMS (EI). Calculated for C9H11BrOS 245.9714. Found 245.9716.
(E)-5-(Bromomethylene)-3-tert-butylthiophen-2(5H)-one 6h. Eluent Hexanes-EtOAc 10
:1; δH (200 MHz; CDCl3) 1.26 (9H, s, t-Bu), 6.54 (1H, d, J 0.9, H1′), 7.56 (1H, d, J 0.9, H4); m/z (EI) 248 (M++2, 88%), 246 (M+, 81) 233 (45), 231 (46), 206 (49), 205 (51), 204 (47), 203 (43), 167 (100), 107 (73), 91 (64).
(Z)-3-Bromo-5-(bromomethylene)thiophen-2(5H)-one 3i. Gradient elution 0–15% EtOAc in hexanes; mp 97–101 °C; δH (200 MHz; CDCl3) 7.15 (1H, s, H1́), 7.65 (1H, s, H4); δC (75 MHz; CDCl3) 113.8, 125.6, 141.9, 145.7, 186.3; m/z (EI) 272 (M++4, 6%), 270 (M++2, 12), 268 (M+, 6), 135(100); HRMS (EI). Calculated for C5H2Br2OS 267.8193. Found 267.8188.
(E)-3-Bromo-5-(bromomethylene)thiophen-2(5H)-one 6i. Gradient elution 0–15% EtOAc in hexanes; δH (200 MHz; CDCl3) 6.73 (1H, s, H1′), 8.09 (1H, s, H4); δC (75 MHz; CDCl3) 108.5, 127.4, 136.2, 143.4, 187.2; m/z (EI) 272 (M++4, 51%), 270 (M++2, 100), 268 (M+, 49), 244 (5), 242 (11), 240 (5), 191 (39), 189 (37), 163 (329, 161 (31), 82 (41).
(Z)-5-(Bromomethylene)-3-(thiophen-2-yl)thiophen-2(5H)-one 3j. Gradient elution 0–15% EtOAc in hexanes; mp 81–83 °C; δH (200 MHz; CDCl3) 7.07(1H, s, H1′), 7.07 (1H, dd, J 5.1, 3.7), 7.41 (1H, dd, J 5.1, 0.9Hz), 7.53 (1H, s, H4), 7.71 (1H, dd, J 3.7, 0.9); δC (75 MHz; CDCl3) 111.3, 128.0, 128.1, 128.8, 129.0, 132.4, 136.9, 142.3, 190.8; m/z (EI) 274 (M++2, 100%), 272 (M+, 93), 246 (33), 244 (31), 165 (55), 121 (80); HRMS (EI). Calculated for C9H5BrOS2 271.8965. Found 271.8970.
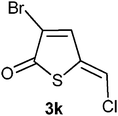
(Z)-3-Bromo-5-(chloromethylene)thiophen-2(5H)-one 3k. Tetrabutylammonium chloride (129 mg, 0.46 mmol) was added to a solution of compound 3i (48 mg, 0.18 mmol) in deuterated chloroform (1 mL). The mixture was stirred for 24 h before it was directly purified by flash column chromatography using hexane/EtOAc 8
:1 for elution; yield 34 mg (86%); δH (200 MHz; CDCl3) 6.87 (1H, s, H1́), 7.66 (1H, s, H4); δC (75 MHz; CDCl3) 123.7, 124.5, 138.8, 145.1, 186.1; m/z (EI) 228 (M++4, 28%), 226 (M++2, 100), 224 (M+, 75), 147 (21), 145 (56), 119 (21), 117 (53),; HRMS. Calculated for C5H2BrClOS 223.8698. Found 223.8700.
5-(Iodomethylene)thiophen-2(5H)-one 3l. Compound 3b (108 mg, 0.56 mmol) in acetone (2 mL) was added dropwise to a solution of sodium iodide (530 mg, 3.5 mmol) in acetone (3 mL). The mixture was stirred at room temperature for 3d before ether (20 mL) and water (20 mL) were added and the product extracted into ether. The crude product was purified by flash chromatography using hexane/EtOAc 5
:1 for elution; yield 97 mg (73%); δH (200 MHz; CDCl3) 6.64 (1H, d, J 6.0, H3), 7.42 (1H, s, H1′), 7.44 (1H, d, J 6.0, H4); δC (75 MHz; CDCl3) 87.2, 133.7, 147.9, 151.7, 193.4; m/z (EI) 238 (M+, 100%), 127 (6), 111 (29), 83 (31); HRMS (EI). Calculated for C5H3IOS 237.8949. Found 237.8942.
5-(Azidomethylene)thiophen-2(5H)-one 3m. Sodium azide (65 mg, 1.0 mmol) was added at 0 °C to a solution of compound 3b (35 mg, 0.22 mmol) in N,N-dimethylformamide (1 mL). The mixture was stirred for 20 min before it was poured into a mixture of ether and water. The product was extracted into ether, washed with brine, dried (MgSO4) and evaporated; yield 30 mg (89%); δH (200 MHz; CDCl3) 6.33 (1H, d, J 6.0, H3), 7.03 (1H, s, H1′), 7.51 (1H, d, J 6.0, H4); m/z (EI) 153 (M+, 41%), 127 (6), 125 (13), 98 (93), 70 (100). 69 (67); HRMS (EI). Calculated for C5H3N3OS 152.9997. Found 152.9993.
5-(Thiocyanatomethylene)thiophen-2(5H)-one 3n. Compound 3b (0.200 g, 1.05 mmol) in acetone (0.5 mL) was added dropwise to a solution of ammonium thiocyanate (0.239 g, 3.14 mmol) in acetone (2.5 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 3 h and then slowly heated to room temperature over the next 1.5 h. The reaction mixture was diluted with ether (20 mL) and washed with water (2 × 10 mL). The combined aqueous phases were extracted with ether (2 × 15 mL). The combined organic phases were dried (MgSO4) and the solvents removed in vacuo; yield (0.111 g, 62%); δH (300 MHz; CDCl3) 6.46 (1H, d, J 5.9, H3), 6.84 (1H, s, H1′), 7.57 (1H, d, J 6.0, H4); δC (75 MHz; CDCl3) 107.4, 117.2, 131.9, 144.1, 146.9, 190.8; m/z (EI) 169 (M+, 86%), 141 (100), 97 (28), 71 (46), 45 (46); HRMS (EI). Calculated for C6H3NOS2 168.9656. Found 168.9653.
3-Bromo-5-(thiocyanatomethylene)thiophen-2(5H)-one 3o. Ammonium thiocyanate (21 mg, 0.28 mmol was added in portions to a solution of compound 3i (42 mg, 0.16 mmol) in acetone (1 mL) at 0 °C. The reaction mixture was stirred at room temperature for 30 min before it was diluted with ether (10 mL) and washed with water (2 × 5 mL). The organic phases was dried (MgSO4) and the solvents removed in vacuo. The crude product was purified by flash chromatography using hexane/EtOAc 5
:1 for elution; yield 31 mg (78%); δH (200 MHz; CDCl3) 6.88 (1H, s, H1′), 7.72 (1H, s, H4); δC (75 MHz; CDCl3) 107.3, 117.8, 125.9, 141.5, 145.5, 184.2; m/z (EI) 169 (M+, 86%), 141 (100), 97 (28), 71 (46), 45 (46); HRMS. Calculated for C6H3NOS2 168.9656. Found 168.9653.
(Z)-(5-Oxothiophen-2(5H)-ylidene)methyl acetate 5. Acetyl bromide (1.0 mL, 13.4 mmol) was added dropwise to a solution of 5-formyl-2-methoxythiophene12 (120 mg, 0.8 mmol) in ether (5 mL) at 0 °C. The reaction mixture was stirred for 3 days before it was diluted with ether (50 mL) and washed with water (2 × 10 mL). The organic phase was dried over MgSO4, filtered, and evaporated. The product mixture was purified by flash chromatography using gradient elution (0–20%) EtOAc in hexanes; yield 40 mg (29%); mp 107–110 °C; ir: 3071, 1772, 1658, 1636, 1565 cm−1; δH (200 MHz; CDCl3) 2.26 (3H, s, Me) 6.31(1H d, J 5.9), 7.61(1H, d, J 5.9), 7.91(1H, s); δC (75 MHz; CDCl3) 20.4, 123.2, 129.5, 147.2, 136.1, 166.1, 193.8; m/z (EI) 170 (M+, 15%), 128 (8), 71(11), 43(100); HRMS (EI). Calculated for C7H6O3S 170.0038. Found 170.0042.
(Z)-5-(Bromomethylene)-3-(hydroxymethyl)thiophen-2(5H)-one 9a. Silver triflate (5.81g, 22.6 mmol) was added to a mixture of 3d1 and 3d2 (69
:31) (3.18 g, 12.7 mmol) in 30 mL acetone/water (9:1). The reaction mixture was stirred for 36 h in the dark, diluted with 100 mL ether and washed with NaCl (aq, sat. 2 × 25 mL). The combined aqueous phases were extracted with ether (2 × 25 mL), and the combined organic phases were dried over MgSO4, filtrated and the solvents were removed in vacuo. The reaction mixture was purified by flash column chromatography using gradient elution (0–40% EtOAc in hexanes); yield 1.93g (69%); mp 88–90 °C; δH (200 MHz; CDCl3) 4.44 (2H, d, J 1.2, CH2), 7.07(1H, s, H'), 7.39(1H, t, J 1.2, H4), δC (75 MHz; CDCl3) 58.0, 112.2, 142.1, 142.9, 145.0, 192.8; m/z (EI): 222 (M++2, 77%), 220 (M+, 76), 193(100), 191(98), 141(29), 81(58); HRMS (EI). Calculated for C6H5BrO2S 219.9194. Found 219.9196; IR: 3261, 3063, 1674.
(Z)-5-(Bromoethylene)-3-(hydroxymethyl)thiophen-2(5H)-one 9b. Compound 3e (0.400 g, 1.34 mmol, 1.0 equiv.) was dissolved in acetone:H2O (9
:1). Silver triflate (0.688 g, 2.69 mmol, 2.0 equiv.) was added and the reaction mixture was stirred at room temperature in the dark for 2.5 h. The reaction mixture was diluted with 50 mL ether and washed with brine (3 × 25 mL). The combined aqueous phases were extracted with ether (2 × 25 mL). The combined organic phases were dried over MgSO4 and the solvents removed in vacuo. The crude product was purified by flash chromatography using gradient elution (5–30% EtOAc in hexanes); yield 0.227 g, (72%); δH (200 MHz; CDCl3) 1.43 (3H, d, J 6.5 Hz, CH3), 2.50 (1H, s, OH), 4.70 (1H, q, J 6.5, 6.4, OCH), 7.06 (1H, s, H'), 7.34 (1H, d, J 0.7, H4); δC (75 MHz; CDCl3) 22.0, 64.2, 112.2, 141.0 , 142.6, 149.1, 192.9; m/z (EI) 236 (M++2, 31%), 234 (M+, 30), 221 (48), 219 (52), 193(100),191 (99), 155 (32), 95 (21); HRMS (EI). Calculated for C7H7O4SBr 233.9350. Found 233.9355.
(Z)- and (E)-5-(1-Bromoethylidene)thiophen-2(5H)-one 10a and 11a. Acetyl bromide (34 mg, 0.28 mmol) in deuterated chloroform (0.5 mL) was add dropwise to a solution of 5-acetyl-2-methoxythiophene14 (21 mg, 0.13 mmol) in deuterated chloroform (1 ml) at 0 °C. The mixture was stirred for 24 h at room temperature before another portion of acetyl bromide (0.15 mL) was added. The mixture stirred at room temperature for 3 days min before the solvent was evaporated off and the residue purified by flash column chromatography using hexane/EtOAc 5
:1 for elution.
Z-isomer 10a. 5 mg (19%); δH (200 MHz; CDCl3) 2.68 (3H, s), 6.48 (1H, d, 1H, J 6.0), 7.68 (1H, d, J 6.0); δC (75 MHz; CDCl3) 25.9, 129.2, 132.6, 139.8, 143.3, 194.7; m/z (EI) 206 (M++2, 88%), 204 (M+, 87), 178 (10), 176 (10), 125 (52), 124 (13), 97 (100); HRMS (EI). Calculated for C6H5BrOS 203.9244. Found: 203.9242. E-isomer 11a: 11 mg (45%); δH (200 MHz; CDCl3) 2.27 (3H,s), 6.44 (1H, d, J 6.0), 7.89 (1H, d, J 6.0); δC (75 MHz; CDCl3) 29.4, 123.7, 129.3, 132.8, 148.1, 194.4; m/z (EI) 206 (M++2, 84), 204 (M+, 84), 178 (10), 176 (10), 125 (56), 124 (15), 97 (100); HRMS (EI). Calculated for C6H5BrOS 203.9244. Found 203.9250.
(Z)- and (E)-5-(Bromo(phenyl)methylene)thiophen-2(5H)-one 10c and 11c. Acetyl bromide (1.5 mL) was added to a solution of 5-benzoyl-2-methoxythiophene15 (100 mg, 0.46 mmol) in dichloromethane (2 mL). The mixture was stirred at room temperature for 8 d and at reflux for 4 h, before the solvent was evaporated off and the residue purified by flash column chromatography using hexane/EtOAc 5
:1 for elution.
Z-isomer 10c. Yield 38 mg (31%); δH (200 MHz; CDCl3) 6.48 (1H, d, J 6.0), 7.43 (5H, Ph), 7.49 (1H, d, J 6.0); δC (75 MHz; CDCl3) 128.6, 129.3, 129.9, 130.3, 132.8, 137.1, 140.9, 145.6, 193.5; m/z (EI) 268 (M++2, 83), 266 (M+, 81), 240 (7), 238 (7), 187 (100), 159 (39), 115 (70). HRMS (EI). Calculated for C11H7BrOS 265.9401. Found 265.9396.
E-isomer 11c. Yield 17 mg (14%); δH (200 MHz; CDCl3) 6.53 (1H, d, J 6.0), 7.3–7.4 (m, 3H, Ph), 7.5–7.6 (2H, m, Ph), 8.12 1H, (d, J 6.0); δC (75 MHz; CDCl3) 128.5, 129.4, 130.5, 132.0 136.0, 138.8, 145.6, 148.5, 195.8; m/z (EI) 268 (M++2, 74), 266 (M+, 73), 240 (7), 238 (7), 187 (100), 159 (36), 158 (14), 115 (71
HRMS (EI). Calculated for C11H7BrOS 265.9401. Found 265.9398.
(Z)-N-((5-Oxothiophen-2(5H)-ylidene)methyl)-N-phenylacetamide 10d. Acetyl bromide (61 mg, 0.50 mmol) in deuterated chloroform (1 mL) was add dropwise to a solution of 1d (74 mg, 0.34 mmol) in deuterated chloroform (1 ml) at 0 °C. The mixture was stirred for 30 min before the solvent was evaporated off and the residue purified by flash column chromatography using hexane/EtOAc 2
:1 for elution; yield 50 mg (60%); δH (200 MHz; CDCl3) 1.94 (3H, s), 6.05 (1H, d, J 6.2), 7.2–7.3 (2H, Ar), 7.4–7.5 (3H, Ar), 7.58 (1H, d, J 6.2 Hz), 8.06 (1H, s); δC (75 MHz; CDCl3) 22.9, 119.5, 124.3, 129.6, 130.3, 137.3, 170.1, 196.0; m/z (EI) 245 (M+, 21%), 203 (100), 202 (29), 175 (7), 174 (10), 130 (7), 104 (10), 77 (27); HRMS (EI). Calculated for C13H11NO2S 245.0511. Found 245.0518.
(Z)-5-(2,2-Dibromoethylidene)thiophen-2(5H)-one 10e. Acetyl bromide (1 mL) was added to a solution of 1c (230 mg, 0.78 mmol) in deuterated chloroform (1 mL) at 0 °C. The mixture was stirred for 5 h at room temperature before it was evaporated. The residue was dissolved in ether, washed with NaHCO3 and brine. The organic phases were dried over MgSO4, filtered, and evaporated. The crude product purified by flash column chromatography using hexane/EtOAc 5
:1 for elution; yield 105 mg (47%); δH (200 MHz; CDCl3) 6.23 (1H, d, J 10.4), 6.43 (1H, d, J 6.4), 6.69 (1H, d, J 10.4), 7.56 (1H, d, J 6.4); δC (75 MHz; CDCl3) 34.5, 132.6, 133.2, 137.7, 149.2, 191.9; m/z (EI) 286 (M++4, 3%), 284 (M++2, 5), 282 (M+, 3), 205 (100) 203 (98), 177 (15), 1275 (15), 96 (33); HRMS (EI). Calculated for C6H4Br2OS 281.8350. Found 281.8358.
References
- J. W. Costerton, K. J. Cheng, G. G. Geesey, T. I. Ladd, J. C. Nickel, M. Dasgupta and T. J. Marrie, Annu. Rev. Microbiol., 1987, 41, 435–464 CrossRef CAS.
- A. Jayaraman and T. K. Wood, Annu. Rev. Biomed. Eng., 2008, 10, 145–167 CrossRef CAS.
- B. L. Bassler, Curr. Opin. Microbiol., 1999, 2, 582–587 CrossRef CAS.
- (a) D. Ren, L. A. Bedzyk, P. Setlow, D. F. England, S. Kjelleberg, S. M. Thomas, R. W. Ye and T. K. Wood, Appl. Environ. Microbiol., 2004, 70, 4941–4949 CrossRef CAS; (b) M. Hentzer, H Wu, J. B. Andersen, K. Riedel, T. B. Rasmussen, N. Bagge, N. Kumar, M. A. Schembri, Z. Song, P. Kristoffersen, M. Manefield, J. W. Costerton, S. Molin, L. Eberl, P. Steinberg, S. Kjelleberg, N. Høiby and M. Givskov, EMBO J., 2003, 22, 3803–3815 CrossRef CAS; (c) T. Defoirdt, C. M. Miyamoto, T. K. Wood, E. A. Meighen, P. Sorgeloos, W. Verstraete and P. Bossier, Environ. Microbiol., 2007, 9, 2486–2495 CrossRef CAS; (d) H. Wu, Z. Song, M. Hentzer, J. B. Andersen, S. Molin, M. Givskov and N. Høiby, J. Antimicrob. Chemother., 2004, 53, 1054–1061 CrossRef CAS; (e) M. B. Jones, R. Jani, D. Ren, T. K. Wood and M. J. Blaser, J. Infect. Dis., 2005, 191, 1881–1888 CrossRef CAS; (f) J. C. Janssens, H. Steenackers, S. Robijns, E. Gellens, J. Levin, H. Zhao, K. Hermans, D. De Coster, T. L. Verhoeven, K. Marchal, J. Vanderleyden, D. E. De Vos and S. C. De Keersmaecker, Appl. Environ. Microbiol., 2008, 74, 6639–6648 CrossRef CAS.
- Y. Han, S. Hou, K. A. Simon, D. Ren and Y. Y. Luk, Bioorg. Med. Chem. Lett., 2008, 18, 1006–1010 CrossRef CAS.
- W. K. Goh, G. Iskander, D. S. Black and N. Kumar, Tetrahedron Lett., 2007, 48, 2287–2290 CrossRef CAS.
- (a) H. J. Jakobsen, E. H. Larsen and S. O. Lawesson, Tetrahedron, 1963, 19, 1867–1882 CrossRef CAS; (b) W. R. Biggerstaff and K. L. Stevens, J. Org. Chem., 1963, 28, 733–736 CrossRef CAS; (c) H. Halvorsen, H. Hope and J. Skramstad, Synth. Commun., 2007, 37, 1167–1177 CrossRef CAS.
- (a) J. E. Schachtner, H.-D. Stachel, K. Polborn and T. Anke, Eur. J. Med. Chem., 1998, 33, 309–319 CrossRef CAS; (b) C. Bongards and W. Gärtner, Eur. J. Org. Chem., 2007, 5749–5758 CrossRef CAS.
- K. Takahashi, T. Sakae and K. Takase, Chem. Lett., 1980, 179–182 CrossRef CAS.
- (a) M. Givskov, R. deNys, M. Manefield, L. Gram, R. Maximilien, L. Eberl, S. Molin, P. D. Steinberg and S. Kjelleberg, J. Bacteriology, 1996, 178, 6618–6622 CAS; (b) R. de Nys, M. Givskov, N. Kumar, S. Kjelleberg and P. D. Steinberg, Progress in Molecular and Subcellular Biology, 2006, 55–86 CAS.
- A. D. Wright, R. de Nys, C. K. Angerhofer, J. M. Pezzuto and M. Gurrath, J. Nat. Prod., 2006, 69, 1180–1187 CrossRef CAS.
- I. M. Downie, M. J. Earle, H. Heaney and K. F. Shuhaibar, Tetrahedron, 1993, 49, 4015–4034 CrossRef CAS.
- J. M. Lansinger and R. C. Ronald, Synth. Commun., 1979, 9, 341–349 CrossRef CAS.
- J. Sicé, J. Am. Chem. Soc., 1953, 75, 3697–3700 CrossRef.
- M. Fiorenza, A. Ricci, S. Giuseppe, G. Pirazzini, C. Eaborn and J. G. Stamper, J. Chem. Soc., Perkin Trans. 2, 1978, 1232–1239 RSC.
- S. Clementi and G. Marino, Tetrahedron, 1969, 25, 4599–4603 CrossRef CAS.
- J. Boukouvalas and Y. Cheng, Tetrahedron Lett., 1998, 39, 7025–7026 CrossRef CAS.
- E. Diez and M. Rico, J. Mol. Spectrosc., 1971, 37, 131–146 CrossRef CAS.
- T. Benneche, G. Herstad and A. Aa. Scheie, unpublished results .
- J. Lönn-Stensrud, T. Benneche and A. Aa. Scheie, Proceedings from ICAR meeting 2010, in press.
- H. Ceri, M. E. Olson, C. Stremick, R. R. Read, D. Morck and A. Buret, J. Clin. Microbiol., 1999, 37, 1771–1776 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00254f |
| This journal is © The Royal Society of Chemistry 2011 |