Cyclopropenium-activated Beckmann rearrangement. Catalysis versus self-propagation in reported organocatalytic Beckmann rearrangements†
Christine M.
Vanos
and
Tristan H.
Lambert
*
Department of Chemistry, 3000 Broadway, New York, NY 10027, USA. E-mail: TL2240@Columbia.edu; Fax: +1 212 932 1289; Tel: +1 212 854 1438
First published on 12th October 2010
Abstract
The concept of cyclopropenium activation has been extended to include dehydrative rearrangements in the context of the Beckmann rearrangement. Geminal dichlorocyclopropenes are shown to rapidly and efficiently convert oximes to amides at room temperature, with reactivity that far surpasses other organic-based promoters. Twelve total examples are provided, including a complex steroidal substrate on preparative scale. Evidence is provided that suggests previously reported organocatalytic Beckmann rearrangements may in fact be self-propagating rather than catalytic.
Aromaticity provides a strong driving force for the generation of cyclopropenium ions via ionization of suitably substituted cyclopropenes.1,2 Our group has discovered that cyclopropenium ions offer a potent and convenient means to promote dehydration reactions, a strategy which we have reported in the context of the nucleophilic substitution of alcohols3 and carboxylic acids (Scheme 1A).4 Importantly, the cyclopropenium ions used in these protocols can be easily prepared in situ from cyclopropenones and are thus readily available and broadly sterically and electronically modifiable.2 Because of this reagent versatility, cyclopropenium ions offer a highly advantageous new platform for synthetic method design, particularly with potential for asymmetric reaction promotion, which is one of our major goals (Scheme 1B). As a key part of our development efforts, we have been investigating the scope of reaction processes for which cyclopropenium activation is effective and provides clear advantage over existing technologies.
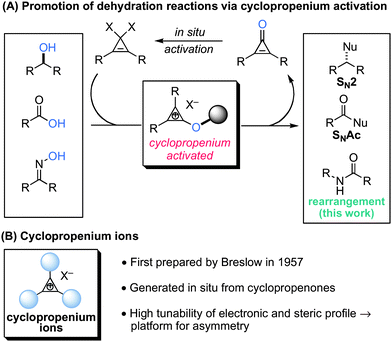 | ||
| Scheme 1 Cyclopropenium activation. | ||
In this regard we became interested in the question of whether cyclopropenium activation could be extended to the promotion of dehydrative molecular rearrangements, such as the Beckmann rearrangement.5 This classic rearrangement of oximes offers a strategically useful means to construct amides and lactams. Traditionally effected by strong Brønsted acid or other common dehydrating agents, recently several organocatalytic methods have been reported.6,7 While effective, these protocols typically required refluxing acetonitrile and Lewis acid co-catalysis6b,c,e or fluorous solvent.6d Arguably, the reagents employed (chloral, cyanuric chloride, tosyl chloride, BOP–Cl) are significantly limited in their suitability for structural modification for the purposes of reactivity enhancement or enantioselective design.
Thus with the goals of (1) further extending the concept of cyclopropenium activation to include dehydrative rearrangements, and (2) developing a new platform for promotion of the Beckmann rearrangement amenable to enantioselection, we decided to investigate whether cyclopropenium ions could serve as an effective means to promote this important transformation. In fact, as described herein, we have found that cyclopropenium activation offers reactivity significantly greater than that of related methods. Importantly, during the course of our studies we have also come to believe there is strong reason to question whether reported organocatalytic methods are actually catalytic or rather merely self-propagating (see below).8
We first sought to determine whether cyclopropenium activation was a viable strategy for the promotion of the Beckmann rearrangement. In fact, when cyclohexanone oxime (1) was treated with 1,1-dichloro-2,3-diphenylcyclopropenone (2) in MeCN at room temperature for 2 h, we obtained, after aqueous workup, a 95% yield of ε-caprolactam (3) (Scheme 2). For comparison, cyanuric chloride in DMF solvent (which presumably forms the Vilsmeier reagent) has been reported9 to require 3 h hours for the conversion of 1 to 3. Obviously, cyclopropenium activation provides an effective means to promote the Beckmann rearrangement.
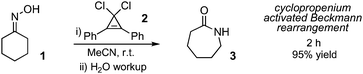 | ||
| Scheme 2 Demonstration of cyclopropenium-activated Beckmann rearrangement. | ||
To further optimize this process, we conducted a screen of substituted dichlorocyclopropenes, which were prepared in situ from the readily available cyclopropenones by simple treatment with oxalyl chloride (Table 1). With acetophenone oxime (4) as the substrate, diphenylcyclopropene 2 was found to induce rearrangement in 70% yield (1H NMR) after 90 min (entry 1). Because we have previously observed a positive correlation between cyclopropenium ion stability and reaction efficiency,4 we next examined the reactivity of a dialkyl-substituted cyclopropene, which are known to possess higher pKR+ values.10 Indeed, we found that the reaction time was reduced and the yield improved with the use of 1,1-dichloro-2,3-diisopropylcyclopropene (6) (entry 2). Further rate enhancement was observed by switching to 4-methoxyphenyl substituents (entry 3). Interestingly, optimal results were observed with the use of either 2,4-xylyl- or mesityl-substituted cyclopropenes 8 or 9, providing N-phenylacetamide 5 in high yield in 20–30 min at room temperature (entries 4 and 5). For comparison, cyanuric chloride in DMF required 6 h to reach completion with this substrate.9 The basis for the superior reactivity of cyclopropenes 8 and 9 is unknown, and warrants further investigation. For reasons of operational convenience, we selected dimesitylcyclopropene 9 as our optimal reagent.
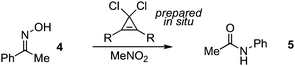
| entry | Rc | mol (%) | T/°C | time/min | % yield | |
|---|---|---|---|---|---|---|
| a Oxalyl chloride (1 equiv. relative to cyclopropenone) was added to the cyclopropenone in MeNO2. After 45 min 4 was added as a solution in MeNO2. b Yield determined by 1H NMR versus Bn2O as an internal standard. c Xyl = 2,4-dimethylphenyl, Mes = 2,4,6-trimethylphenyl. d Isolated yield. | ||||||
| 1 | Ph | 2 | 105 | 21 | 90 | 70 |
| 2 | iPr | 6 | 105 | 21 | 75 | 77 |
| 3 | 4-OMe-Ph | 7 | 105 | 21 | 45 | 77 |
| 4 | Xyl | 8 | 105 | 21 | 20 | 98 |
| 5 | Mes | 9 | 105 | 21 | 30 | 92d |
| 6 | Mes | 9 | 10 | 21 | 30 | 10 |
| 7 | Mes | 9 | 10 | 60 | 30 | 100 |
| 8 | Mes | 9 | 5 | 60 | 90 | 100 |
| 9 | Mes | 9 | 2 | 60 | 8 h | 24 |
Given the observed high reactivity in comparison to cyanuric chloride and the previous report of catalysis with that reagent, we were interested to see whether our cyclopropenium activation strategy could also be rendered catalytic. With this aim, we attempted the rearrangement of oxime 4 using 10 mol% dimesitylcyclopropene, but at room temperature observed only 10% of the desired product 5 (Table 1, entry 6). On the other hand, heating the reaction mixture to 60 °C resulted in quantitative conversion after 30 min (entry 7). Reducing the loading to 5 mol% was also possible, although the reaction time increased to 90 min (entry 8). A loading of 2 mol% produced inferior results (entry 9).
In contemplating the mechanism by which this substoichiometric rearrangement might have occurred, we envisioned two possibilities (Scheme 3). First, we reasoned that upon rearrangement of the cyclopropenium oxime 17, the resulting nitrilium ion 16 could alkylate the expelled cyclopropenone 14 to produce a cyclopropenium imidate 13. This imidate could then undergo exchange with another molecule of oxime substrate 10 to release the amide product 12 and thereby propagate the catalytic cycle (path a). In fact, this hypothesis is directly analogous to the one first proposed by Ishihara and Yamamoto for their cyanuric chloride-catalyzed protocol.6b On the other hand, we recognized another possibility: that nitrilium ion 16 (or the imidoyl chloride 15) might alkylate another molecule of oxime 10 directly, thus giving rise to intermediate 18, which could then undergo Beckmann rearrangement to produce the amide product and regenerate nitrilium 16 (path b). The distinction between this self-propagating mechanism and that of genuine catalysis (path a) is an important one, particularly for contexts in which catalyst control would be essential (e.g. asymmetric kinetic resolution).
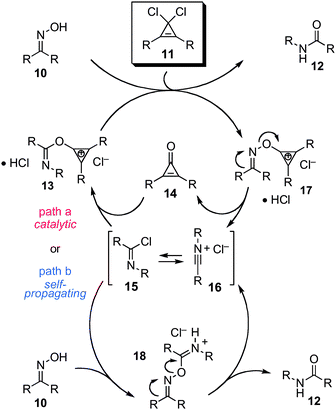 | ||
| Scheme 3 Mechanistic analysis of substoichiometric cyclopropenium-mediated Beckmann rearrangement. | ||
To distinguish between these two possibilities, we first ran a competition experiment between acetophenone oxime 4 and 2,3-dimesitylcyclopropenone 19 for reaction with oxalyl chloride (Scheme 4). In fact, when equimolar amounts of 4 and 19 were combined and treated with one equivalent of oxalyl chloride, we observed only oxalate ester 20 and none of the Beckmann rearrangement product 21, which would be produced if the cyclopropenone had reacted to form dichlorocyclopropene. This result demonstrates that this cyclopropenone is not nucleophilically competitive with oximes. As further evidence, we prepared N-phenylbenzimidoyl chloride (24) (the Beckmann rearrangement product of benzophenone oxime 22), and found that at 50 °C, 10 mol% of this reagent in the presence of HCl effectively promoted the Beckmann rearrangement of benzophenone oxime 22 in high yield (Scheme 5). Obviously, the evidence is heavily in favor of the self-propagating mechanism (path b) rather than catalysis (path a).
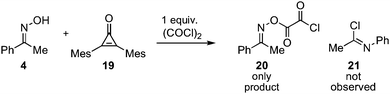 | ||
| Scheme 4 Reactivity comparison between oxime 4 and cyclopropenone 19. | ||
In light of these results, we suspected there was a strong possibility that the reported organocatalytic Beckmann rearrangement protocols that employed cyanuric chloride, tosyl chloride, or BOP–Cl might have also proceeded via self-propagation rather than catalysis. If this were the case, one might reasonably expect to observe during the course of these reactions the presence of imidoyl chloride intermediates (cf.15, Scheme 3), which would not be present if the proposed mechanisms were operative. Furthermore, one would expect to find that imidoyl chlorides could promote rearrangement at rates comparable to those using the promoters themselves. To investigate, we compared the rate of conversion of benzophenone oxime (22) to 23 using imidoyl chloride 24versus cyanuric chloride (25) and found that the reactions proceeded at essentially identical rates (Scheme 5). In addition, we were able to observe by 1H NMR imidoyl chloride 24 in the reaction mixture with cyanuric chloride (25) as promoter. Thus, because the rate of the imidoyl chloride-promoted reaction is comparable to the rate of the purported catalytic reaction and because 24 is demonstrably present during the course of this reaction, we conclude that the reported cyanuric chloride-catalyzed Beckmann rearrangement, at least with this substrate, is reagent initiated and subsequently self-propagating. Although further studies are warranted, based on this finding we believe there is sufficient cause for skepticism regarding the mechanistic claims of reported organocatalytic Beckmann rearrangements.
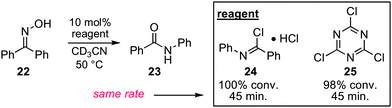 | ||
| Scheme 5 Rate comparison of imidoyl chloride versus cyanuric chloride. | ||
Because imidoyl chlorides may promote oxime rearrangement at elevated temperatures, the true reactivity comparison of various Beckmann reagents must be conducted under conditions in which the imidoyl chloride products are not effective (e.g. at room temperature). In this regard, we reiterate our finding that cyclopropene 8 effects the Beckmann rearrangement of acetophenone oxime (4) in high yield in only 20 min at ambient temperature, noting that cyanuric chloride on its own was ineffective at this temperature, and in DMF solvent has been reported to require 6 h for comparable conversion (Table 2).9

A further investigation of substrate scope has revealed similarly high reactivity for cyclopropenium-activated Beckmann rearrangements (Table 3). (Note that in some cases diphenylcyclopropene 2 was used to aid in chromatographic purification of the products.) Thus several benzophenone (entries 1 and 2) and acetophenone (entries 3–6) oximes were found to undergo rapid rearrangement in good to high yields. In addition to these aryl oximes, dibenzyl, cyclohexyl, and cyclododecyl oximes were also subject to Beckmann rearrangement with high efficiency (entries 7–9). The short reaction time to produce ε-caprolactam is particularly noteworthy, given that the Vilsmeier reagent requires 3 h.9 In addition, we investigated the reaction of cyclohexyl phenyl oxime, which is formed from the corresponding ketone as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E:Z mixture. As expected, exclusive trans migration was observed, leading to a 2:1 mixture of amide products 26 and 27 in high yield (entry 10). This result suggests that oxime isomerization does not occur under these reaction conditions.
:1 E:Z mixture. As expected, exclusive trans migration was observed, leading to a 2:1 mixture of amide products 26 and 27 in high yield (entry 10). This result suggests that oxime isomerization does not occur under these reaction conditions.
| entry | substrate | product | R | time/min | % yield |
|---|---|---|---|---|---|
| a Reactions performed as described in Table 1, footnote a. b Yields determined on isolated and purified products unless otherwise noted. Yields in parentheses correspond to reactions run with 10 mol% cyclopropenone and 10 mol% oxalyl chloride at 80 °C in nitromethane. c Reaction performed at 80 °C in MeCN. d Yield determined by 1H NMR analysis versus Bn2O as an internal standard. | |||||
| 1 |

|

|
Ph | 30 | 98 (97) |
| 2 |

|

|
Mes | 20 | 96 (95) |
| 3 |

|
|
Mes | 20 | 93 (98) |
| 4 |

|

|
Mes | 30 | 80 (99) |
| 5 |

|

|
Mes | 90 | 82 (98) |
| 6 |
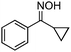
|

|
Ph | 90 | 87 (89) |
| 7 |

|

|
Ph | 120 | 86c (85) |
| 8 |

|
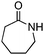
|
Mes | 30 | 96d |
| 9 |

|
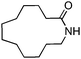
|
Mes | 24 h | 99 (93) |
| 10 |

|

|
Ph | 60 | 99 (99) |
Finally, to demonstrate the compatibility of cyclopropenium activation with a complex molecular setting, we conducted the rearrangement of 3β-acetoxypregnenone oxime 28, which furnished the steroidal amide 29 after 2 h in 90% yield on a 0.9 gram scale (Scheme 6). Alternatively, using 5 mol% cyclopropenone 8 and oxalyl chloride at 80 °C, a 94% yield of 29 could be obtained.
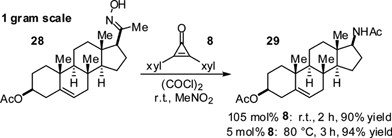 | ||
| Scheme 6 Preparative scale Beckmann rearrangement. | ||
In conclusion, we have extended the concept of cyclopropenium activation to include dehydrative molecular rearrangements, in the context of promotion of the Beckmann rearrangement. Notably, it appears that this strategy offers significantly greater reactivity than other reported organic promoters. In the course of this work we have discovered the intriguing, though as yet unexplained, superior reactivity of xylyl- and mesityl-substituted cyclopropenes. Furthermore, we have provided evidence that previously reported organocatalytic Beckmann rearrangements may instead be merely self-propagating, leaving the development of an organocatalytic Beckmann rearrangement an unsolved challenge. In addition to the pursuit of this goal, our current aims include exploring the possibility of achieving enantioselective Beckmann rearrangements (e.g. asymmetric kinetic resolution) using cyclopropenones as a convenient chiral platform.
THL is grateful for Young Young Investigator Awards from Abbott and Amgen.
Notes and references
- (a) R. Breslow, J. Am. Chem. Soc., 1957, 79, 5318 CrossRef CAS; (b) R. Breslow and C. Yuan, J. Am. Chem. Soc., 1958, 80, 5991 CrossRef CAS.
- For reviews on cyclopropenium ions and cyclopropenones, see: (a) K. Komatsu and T. Kitagawa, Chem. Rev., 2003, 103, 1371 CrossRef CAS; (b) A. W. Krebs, Angew. Chem., Int. Ed. Engl., 1965, 4, 10 CrossRef; (c) Z.-i. Yoshida, Topics in Current Chemistry, 1973, 40, 47 CAS; (d) I. A. D'yakonov and R. R. Kostikov, Russ. Chem. Rev., 1967, 36, 557 Search PubMed.
- B. D. Kelly and T. H. Lambert, J. Am. Chem. Soc., 2009, 131, 13930 CrossRef CAS.
- D. J. Hardee, L. Kovalchuke and T. H. Lambert, J. Am. Chem. Soc., 2010, 132, 5002 CrossRef CAS.
- For a review of the Beckmann rearrangement, see: R. E. Gawley, Org. React., 1988, 35, 1 Search PubMed.
- (a) S. Chandrasekhar and K. Gopalaiah, Tetrahedron Lett., 2003, 44, 755 CrossRef CAS; (b) Y. Furuya, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 11240 CrossRef CAS; (c) M. Zhu, C. Cha, W.-P. Deng and X.-X. Shi, Tetrahedron Lett., 2006, 47, 4861 CrossRef CAS; (d) M. Hashimoto, Y. Obora, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2008, 73, 2894 CrossRef CAS; (e) H.-J. Pi, J.-D. Dong, N. An, W. Du and W.-P. Deng, Tetrahedron, 2009, 65, 7790 CrossRef CAS.
- For selected other examples of Beckmann rearrangements, see: (a) M. Arisawa and M. Yamaguchi, Org. Lett., 2001, 3, 311 CrossRef CAS; (b) Y. Izumi, Chem. Lett., 1990, 19, 2171 CrossRef; (c) H. Kusama, Y. Yamashita and K. Narasaka, Bull. Chem. Soc. Jpn., 1995, 68, 373 CrossRef CAS; (d) T. Mukaiyama and T. Harada, Chem. Lett., 1991, 20, 1653 CrossRef; (e) Y. Kikugawa, C. Tsuji, E. Miyazawa and T. Sakamoto, Tetrahedron Lett., 2001, 42, 2337 CrossRef CAS; (f) R. Anilkumar and S. Chandrasekhar, Tetrahedron Lett., 2000, 41, 5427 CrossRef CAS; (g) A. Laurent, P. Jacquault, J. L. Di Martino and J. J. Hamelin, J. Chem. Soc., Chem. Commun., 1995, 1101 RSC.
- During the preparation of this manuscript, Yadav and coworkers reported that 1,1-dichloro-2,3-diphenylcyclopropene could be used as an “organocatalyst” for the Beckmann rearrangement at elevated temperatures. For the reasons given in this article, we remain skeptical that these reactions occur via cyclopropenium-based catalysis, although obviously further investigation is warranted. V. P. Srivastava, R. P. Patel, Garima and L. D. S. Yadav, Chem. Commun., 2010, 46, 5808–5810 Search PubMed.
- L. D. Luca, G. Giacomelli and A. Porcheddu, J. Org. Chem., 2002, 67, 6272–6274 CrossRef CAS.
- For a large collection of cyclopropenium ion pKR+ values, see: K. Okamoto, K. Takeuchi, K. Komatsu, Y. Kubota, R. Ohara, M. Arima, K. Takahashi, Y. Waki and S. Shirai, Tetrahedron, 1983, 39, 4011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR spectra of equilibration experiments, and full characterization of new compounds. See DOI: 10.1039/c0sc00421a |
| This journal is © The Royal Society of Chemistry 2010 |