Site-selective chemoenzymatic construction of synthetic glycoproteins using endoglycosidases†
Marta
Fernández-González
,
Omar
Boutureira
,
Gonçalo J. L.
Bernardes
,
Justin M.
Chalker
,
Matthew A.
Young
,
James C.
Errey
and
Benjamin G.
Davis
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, United Kingdom. E-mail: Ben.Davis@chem.ox.ac.uk; Fax: +44 (0) 1865 275674; Tel: +44 (0) 1865 275652
First published on 20th October 2010
Abstract
Combined chemical tagging followed by Endo-A catalysed elongation allows access to homogeneous, elaborated glycoproteins. A survey of different linkages and sugars demonstrated not only that unnatural linkages can be tolerated but they can provide insight into the scope of Endo-A transglycosylation activity. S-linked GlcNAc-glycoproteins are useful substrates for Endo-A extensions and display enhanced stability to hydrolysis at exposed sites. O-CH2-triazole-linked GlcNAc-glycoproteins derived from azidohomoalanine-tagged protein precursors were found to be optimal at sterically demanding sites.
Introduction
Glycan display is critical for development and physiology of many living systems.1 Protein glycosylation, in particular is a diverse form of posttranslational modification comprising 50% of cellular proteome and 90% of secreted proteome.2,3 Currently, 70% of the total of therapeutic proteins that are currently in clinical trials are glycoproteins.4 Natural glycoproteins exist as mixtures of glycoforms—same peptide backbone but different glycosylation pattern and site—making isolation of well-defined glycoproteins complicated.5 This results in a lack of detailed structural and functional studies on the specific role of glycans. Moreover, since certain glycoforms are more active than others,6 access to specific uniform glycoforms can confer significant therapeutic advantages.Recombinant expression systems, chemoenzymatic and chemical methods have all emerged as powerful techniques that can resolve the supply issues of homogeneous glycoprotein production.7–12 One powerful approach is to remodel a glycoprotein enzymatically: the initial heterogeneous glycoform mixture is treated with endoglycosidase (“Endo”) to trim off the variable portions of the attached oligosaccharides, yielding a single protein glycoform with N-acetylglucosamine (GlcNAc) residue(s) at N-linked glycosylation site(s) (Route A). Subsequent enzyme-mediated glycosylation of the remaining single GlcNAc residues can then produce a homogenous sample of a desired glycoprotein (Scheme 1, Route A).10,13–16 Endo-β-N-acetylglucosaminidases hydrolyze the glycosidic bond in the N,N′-diacetylchitobiose core of N-linked glycans. Some of them, such as Endo-A17 and Endo-M18 also possess significant transglycosylation activity and have been used, for example, to remodel the N-glycoprotein RNaseB.19,20 Importantly, the efficiency of glycosylation has been valuably improved21–26 by the use of sugar oxazolines as donor substrates and mutant Endo enzymes. Very recently the re-engineering of bacterial glycosylation has been usefully combined with remodeling to extend this method.16 So far, these methodologies have been applied to a limited number of proteins with pre-existing, biologically-defined glycosylation sites, which in turn requires the successful incorporation and recognition of glycosylation peptide consensus motif (NxS/T). In some cases this motif may not be recognised, leading to failed or incomplete glycan incorporation and lowered yields.16
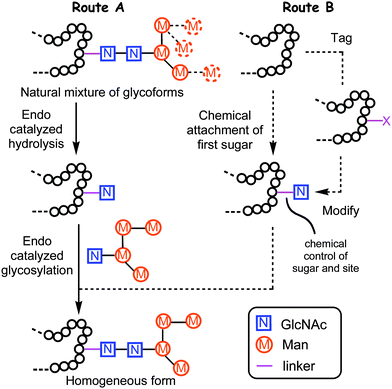 | ||
| Scheme 1 Existing remodeling (A) approach compared with chemoenzymatic (B) strategy, disclosed here, for endoglycosidase-catalysed protein glycosylation. | ||
The power of the Endo-catalysed approach would therefore be enhanced by combination with methods that would allow more free-ranging glycosylation site control and the ability to incorporate unnatural or altered motifs. In this way, a system can be envisaged that would allow Endo-catalysed glycosylation on proteins in which (a) the first GlcNAc (or other) residue that is required had been positioned with complete freedom and (b) the use of other glycans. This would also allow a broad exploration of Endo substrate tolerance in glycoprotein construction and the possible discovery of more efficient substrates and more stable products.
We describe here a method (Scheme 1, Route B) that achieves this using chemical positioning of first sugars. This combined chemoenzymatic approach consists of site-selective convergent chemical glycosylation of a protein scaffold with a single GlcNAc (or other sugars) followed by Endo-catalysed glycosylation and allows greater flexibility.27 The strategy brings with it the potential to use sugars other than GlcNAc as glycosylation acceptors, as well as linkages beyond the δ-amide of Asn. Such ‘unnaturally’ linked acceptors for Endo-catalysed glycosylation have been explored in short peptides28,29 and may bring with them the potential for enhanced yield as a result of reduced product hydrolysis by Endo. We not only use this method here to map the tolerance and efficiency of Endo but use it to discover unnatural glycoprotein linkages that are more stable (and hence more efficiently formed) than the natural.
Results and discussion
We have previously described a number of ‘tag-and-modify’30 methods that allow efficient site-selective glycoconjugation of proteins with single sugars that could in principle act as acceptors in Endo-catalysed glycosylations suggested by Scheme 1B. Here we combine such site-selective glycoconjugation (for the introduction of a first sugar unit into a given protein site) with regio- and stereo-selective Endo-A-catalysed chemoenzymatic extension. To evaluate which potential linkages and sugars could undergo efficient glycosylation we surveyed a number of structures, all of which could be chemically incorporated into proteins in a site-selective manner (Table 1). These included triazole-linked proteins (accessible from azide-(Aha) or alkyne-(Hpg) tagged proteins),31 thioether-linked proteins (accessible from enamide-(Dha) tagged proteins),32 selenenylsulfide33- and disulfide34-linked (accessible from thiol(Cys)-tagged). To create a protein with a unique protein tag, the position of which may be controlled, mutation of a wild-type protein gene sequence may be necessary to allow reassignment of, e.g. Cys or Met, codons for the eventual creation of the tags.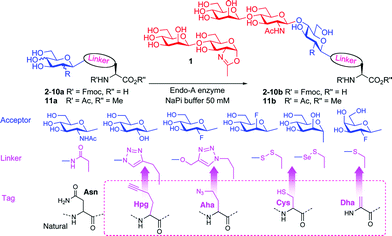
| Entry | Acceptor | 1/μmol | Endo-A [mU/g] | Conda | t mc min | 2–11b Conv [%] |
|---|---|---|---|---|---|---|
| a Conditions: 0.2 μmol acceptor, NaPi buffer (50 mM) and A: pH 6.0, c 0.2 mM, 27 °C; B: pH 7.0, c 0.2 mM, 27 °C; C: pH 7.0, c 2 mM, 21 °C; D: pH 7.0, c 0.2 mM, 21 °C. b t mc: time of maximal conversion. | ||||||
| 1 |
|
4 × 0.6 | 120 | A | 29 | 25 |
| 2 |
|
4 × 0.6 | 70 | A | 48 | 78 |
| 3 |
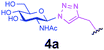
|
0.6 + 0.3 | 20 | A | 26 | 40 |
| 4 | 5 × 0.6 | 110 | A | 48 | 83 | |
| 5 | 5 × 0.6 | 110 | B | 48 | 75 | |
| 6 | 0.6 | 30 | C | 1 | 74 | |
| 7 |
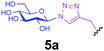
|
0.6 | 30 | D | 48 | 7 |
| 8 | 0.6 | 30 | C | 4 | 50 | |
| 9 |
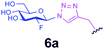
|
1.9 | 60 | D | 120 | 28 |
| 10 | 0.6 | 30 | C | 3 | 34 | |
| 11 |
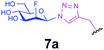
|
0.6 | 30 | C | 5 | 33 |
| 12 |
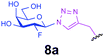
|
0.6 | 30 | C | 24 | 0 |
| 13 |
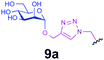
|
0.6 | 30 | C | 24 | 0 |
| 14 |
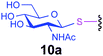
|
2.0 | 30 | C | 20 | 89 |
| 15 |

|
0.6 | 30 | C | 0.5 | 75 |
| 16 | 0.6 | 30 | E | 0.5 | 98 |
Endo-A elongation studies on amino acids and peptides
Representative amino acid models bearing these different linkages with various sugar moieties were prepared and Endo-catalysed glycosylation activity surveyed (Table 1). Disaccharide oxazoline 1 was chosen as donor because it is a minimal and broadly recognised (by both Endo-A and M) substrate in glycosylation.26,35 A wide tolerance was observed for acceptor-linkage types beyond the natural GlcNAc-Asn motif 2a. Best conversions were observed for GlcNAc substrates 3a, 4a, 10a and 11a; Asn-linked GlcNAc 2a, the natural linkage gave the lowest conversion of these GlcNAc containing substrates (Table 1, entry 1). Importantly, these results demonstrated that unnatural linkages could prove better in glycosylations: S- (in 10a), SS- (in 11a) and triazoles (in 3–7a) all proved superior to natural Asn-linked 2a. Interestingly, Endo-A glycosylation activity was also observed in cases where the natural GlcNAc moiety was replaced by unnatural sugars such as 2-deoxy-2-fluoro sugars (Table 1, entries 9–11).Having shown the potential of unnatural linkages and sugars, the convergent, chemoenzymatic strategy outlined in Scheme 1, Route B was demonstrated on peptide substrates. Fragments of cancer antigen protein gp9036 bearing alkyne or azide tags, and fragments of subtilisin Bacillus lentus (SBL) bearing a Cys-tag were assembled using Fmoc chemistry (Scheme 2). GlcNAc moiety was then incorporated convergently using Cu(I)-catalysed [3 + 2] cycloaddition to give 12, 13a,37,38 and in the case of Cys-peptide 14a, by conversion of Cys to Dha using O-mesitylenesulfonylhydroxylamine (MSH)32 followed by conjugate addition of GlcNAc-SH. Subsequent Endo-A glycosylation gave the corresponding elaborated glycopeptides 12–14b. Consistent with the prior observations on amino acid models, S- and triazole also proved to be efficient substrate linkages in peptides: >98% conversions of 13a and 14a were observed after 2 and 7 h, respectively. Lower conversion (70%) was observed in the case of triazole-glycopeptide 12a (from alkynyl tag Hpg).
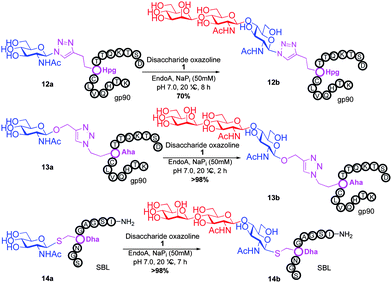 | ||
| Scheme 2 Endo-A catalysed transglycosylation of glycopeptides 12–14; see ESI† for full details. | ||
Chemoenzymatic synthesis of glycoproteins
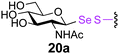
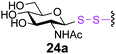
To investigate the important transfer of this chemoenzymatic approach to glycoproteins, we built a series of glycoprotein substrates. As a first substrate class, we explored S-, SeS- and SS-linked glycoproteins. A range of S-linked glycoproteins (15–19a) were prepared by conjugation of glycosyl thiols (GlcNAc, Glc, Gal, Glc2F, Man2F) to a Dha-tagged protein32 and their compatibility with Endo-A transglycosylation studied. We also prepared selenenylsulfide33- (20–23a) and disulfide34- (24a) linked glycoproteins, accessed from Cys-tagged proteins. A wide-ranging activity and tolerance was observed with GlcNAc-proteins in reactions catalyzed by Endo-A. 15, 20 and 24a proved most efficient (70->95%); glycosylation of S-linked 15a afforded glycoprotein 15b in >95% after 6 h. Glc-proteins 16 and 18a, were also recognized by Endo-A as acceptors, although conversions were lower (<35%). Only Gal-tagged protein 17a bearing an unreactive39,40 axial hydroxyl group at C-4 (Table 2, entry 3) gave no transglycosylated protein product, consistent with expected β(1 → 4) glycosylation.21 Importantly, since SBL is a protease, its activity is easily assayed to demonstrate that the protein is not denatured during the installation of GlcNAc or the subsequent Endo-A catalyzed extension. Indeed, SBL-S-GlcNAc 15a and the product of Endo-A modification 15b were both active proteases (see ESI†). This result reflects the mild conditions used throughout the reaction sequence and demonstrates that this tag-and-modify approach is suitable for the synthesis of fully functional glycoproteins.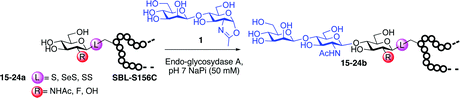
| Entrya | Acceptor | [15–24a]/mg mL−1 | t/h | Max. conv [%] |
|---|---|---|---|---|
| a See ESI† for full details. | ||||
| 1 |

|
1.0 | 6 | 15b [>95] |
| 2 |
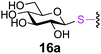
|
0.29 | 4 | 16b [35] |
| 3 |
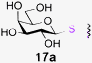
|
0.29 | 6 | 17b [0] |
| 4 |
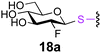
|
0.4 | 24 | 18b [10] |
| 5 |
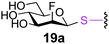
|
0.4 | 24 | 19b [25] |
| 6 |
|
0.29 | 2 | 20b [80] |
| 1.0 | 2 | 20b [89] | ||
| 7 |
|
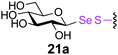
0.29 | 2 | 21b [26] |
| 1.0 | 2 | 21b [28] | ||
| 8 |
|
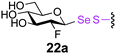
0.5 | 2 | 22b [<10] |
| 9 |
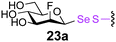
|
0.29 | 2 | 23b [63] |
| 0.43 | 2 | 23b [55] | ||
| 1.0 | 2 | 23b [54] | ||
| 10 |
|
1.0 | 6 | 24b [70] |
The potential to incorporate fluorinated glycans into proteins gives an unnatural motif that not only might modulate Endo-associated kinetics but is a possible strategy for labeling proteins.41 In this context disulfide-forming glyco-PTS42 reagents have been used to introduce PET-reagent FDG (2-fluoro-2-deoxy-Glc) into short peptides.43 We were pleased to see that the Endo-catalysed glycosylation was successful for both 19FDG as an acceptor and its C-2 epimer the 2-F-Man variant (Table 2, entries 4, 5, 8 and 9). Surprisingly, Man-2F-proteins 19 and 23a were better acceptor substrates than corresponding Glc-2F-proteins 18 and 22a (yields up to 63%) and even better in some cases than corresponding Glc-proteins e.g., 21a and 23a (Table 2, entries 7 and 9). This further highlighted the striking promiscuity of Endo discovered with this approach. However, it should be noted that the conversion times established here for Endo-catalysed reactions make this method more compatible with NMR-labeling (19F) than for example radio/PET (18F), given the short half life of 18F.41
To probe the effects of linker upon transglycosylation activity and kinetics, three synthetic glycoproteins bearing different linkages (S-, SS- and SeS-) but identical acceptor sugar GlcNAc were compared (Fig. 1). These gave a clear insight into the higher transglycosylation efficiencies observed for S-linked glycoproteins. Thus, SeS-glycoproteins were not only faster acceptors for Endo-A glycosylation than S- and SS-substrates (Fig. 1) but also for the subsequent hydrolysis of product. Such undesirable hydrolytic behaviour has hampered other Endo-catalysed approaches.14,15 In contrast, S-glycoprotein product 15b was stable and formed with full conversion after 6 h; the corresponding SeS-glycoprotein 20b was hydrolyzed by 60% back to 20a in the same period (Fig. 1).
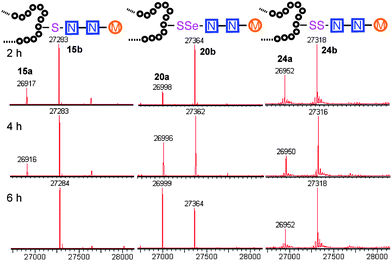 | ||
| Fig. 1 Comparison of linkage type: time course of Endo-A catalysed glycosylation with S-, SeS-, and SS-linked SBL-GlcNAc. | ||
Finally, we set out to compare the S- and triazole linker effects directly. Accordingly, three glycoproteins were constructed that differ only by the linkage bearing the GlcNAc acceptor (25–28a). A model protein, Np276 from Nostoc punctiforme,44 was used to make this comparison. Aha, Hpg, and Cys were incorporated into position 61 as ‘tagged’ proteins or precursors of tagged proteins. This required the construction of appropriate mutant gene sequences to allow site-selective tag positioning (Table 3). GlcNAc was subsequently attached to these tags as follows. Triazole-synthesis chemistry from Aha (azide tag) and Hpg (alkyne tag) gave 25a and 26a, respectively. Conversion of Cys61 to dehydroalanine as a tag and conjugate addition of GlcNAc thiol provided 27a. Position 61 in Np276 was more hindered than 156 in SBL and this was reflected in the longer reaction times required for complete conjugation (see ESI†).
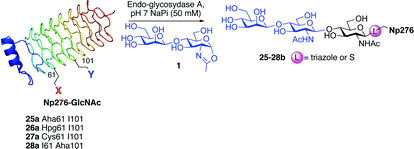
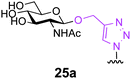
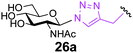

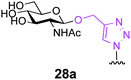
These three differently-linked GlcNAc-ylated protein variants allowed direct comparison at a single site in one protein of triazole- and thioether-linked acceptors. We considered such a hindered site an alternative testing ground for the relative efficiency of each linker in Endo-A catalysed transglycosylations. Interestingly, while the thioether-linked GlcNAc on SBL derivative 15a was an efficient acceptor for the Endo-A catalysed glycosylation, the same thioether-linked sugar at position 61 of Np276 was a poor acceptor and provided no detectable product after 6 h. In contrast, substrate 25a, the O-triazole-linked GlcNAc variant derived from Aha at the same position, did show activity and 40% conversion to the glycosylated product was observed even at this hindered site. No product was observed with the Hpg-derived N-triazole-linked GlcNAc variant 26a. From these results, we can draw the following conclusions. Firstly, it appears that for hindered sites such as position 61 in Np276, the Aha-derived triazole, containing a hydroxymethyl spacer, is superior to both the thioether-linked GlcNAc and the Hpg-derived triazole variants. One factor to account for this difference is simply the relative accessibility of each linker: the Aha derived triazoles are more exposed from the protein surface than the thioether derived from Dha. The triazole motif in 25a is also more spaced from the GlcNAc residue as compared with 26a; this difference is apparently sufficient to lead to 40% conversion for 25a and yet no reaction for 26a. The position on the protein surface is also critical. In our next experiment (entry 4), by moving the Aha azide tag to a more accessible position 101, both the initial installation of GlcNAc and the subsequent Endo-A catalysed transglycosylation were more efficient. Indeed, 28a was then completely consumed in an Endo-catalyzed reaction with 1 after only 4 h.
Conclusions
We have demonstrated a combined chemoenzymatic approach (Scheme 1, Route B) allowing convergent protein glycosylation. A survey of an array of triazole-, S-, SeS-, SS-linked glycoamino acids, glycopeptides and glycoproteins in Endo-A catalysed glycosylation led to discovery of S-glycoproteins as privileged substrates for enzymatic extension, provided the acceptor is in an unhindered site. In the cases where the S-GlcNAc protein acceptor is unhindered, full conversion to the enzymatic extension product is observed without hydrolysis. This result reinforces a longstanding interest in S-glycoconjugate synthesis45 due to their enhanced resistance to chemical46 and enzymatic hydrolysis47 when compared to natural counterparts. This method allows greater freedom of glycosylation site and sugar identity whilst also beneficially reducing product hydrolysis. For more hindered sites, the choice of a triazole-linkage derived from an Aha/azide tag may allow Endo-A catalysed modification where thioether derivatives from Cys fail (Table 3). Together, these results demonstrate key factors that must be considered for the Endo-catalysed method to be expanded to different sites, proteins and glycan acceptors. This method might allow improvements in biotechnologically valuable protein systems, such as antibodies, that have proven to be difficult substrates using other methods that can give lower yields (e.g., 5%–∼40%)16 for glycan incorporation. The ease of assembly of S- and triazole-linked glycoproteins and Endo-A extension makes this a powerful method for the production of more complex homogeneous synthetic glycoproteins.Experimental procedures
Typical procedure for the chemoenzymatic transglycosylation: to a solution of SBL-GlcNAcS156 15a (200 μL of 1 mg mL−1, 0.007 μmol) in sodium phosphate buffer (50 mM solution, pH 7.0), disaccharide oxazoline 1 (600 μg, 1.642 μmol) and Endo-A (10 milliunits) were added and the mixture was placed on an end-over-end rotator at 22 °C. The reaction was analyzed directly by LC-MS.Acknowledgements
We thank Xunta de Galicia, Spain (MFG), EC (Marie Curie IEF, OB), International AIDS Vaccine Initiative (MFG, GJLB), FCT-Portugal and Royal Society of Chemistry's JWT Jones Travelling Fellowship (GJLB), Rhodes Trust and NSF (JMC), BBSRC and UCB-Celltech (MAY) for financial support and M. K. Patel for technical assistance. We also thank Profs L-X. Wang and J.S. Blanchard for kindly providing pGEX-2T/Endo-A and pET28d/Np276 plasmids. BGD is a recipient of a Royal Society Wolfson Merit Award and is supported by an EPSRC LSI Platform grant.Notes and references
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS.
- R. Apweiler, H. Hermjakob and N. Sharon, Biochim. Biophys. Acta, 1999, 1473, 4–8 CrossRef CAS.
- E. Jung, A.-L. Veuthey, E. Gasteiger and A. Bairoch, Proteomics, 2001, 1, 262–268 CrossRef CAS.
- N. Sethuraman and T. A. Stadheim, Curr. Opin. Biotechnol., 2006, 17, 341–346 CrossRef CAS.
- P. M. Rudd, H. C. Joao, E. Coghill, P. Fiten, M. R. Saunders, G. Opdenakker and R. A. Dwek, Biochemistry, 1994, 33, 17–22 CrossRef CAS.
- T. Suzuki, K. Kitajima, S. Inoue and Y. Inoue, Glycoconjugate J., 1995, 12, 183–193 CrossRef CAS.
- T. U. Gerngross, Nat. Biotechnol., 2004, 22, 1409–1414 CrossRef CAS.
- M. J. Betenbaugh, N. Tomiya, S. Narang, J. T. A. Hsu and Y. C. Lee, Curr. Opin. Struct. Biol., 2004, 14, 601–606 CrossRef CAS.
- M. Kowarik, S. Numao, M. F. Feldman, B. L. Schulz, N. Callewaert, E. Kiermaier, I. Catrein and M. Aebi, Science, 2006, 314, 1148–1150 CrossRef CAS.
- C. S. Bennett and C.-H. Wong, Chem. Soc. Rev., 2007, 36, 1227–1238 RSC.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS.
- G. J. L. Bernardes, B. Castagner and P. H. Seeberger, ACS Chem. Biol., 2009, 4, 703–713 CrossRef CAS.
- K. Witte, P. Sears, R. Martin and C.-H. Wong, J. Am. Chem. Soc., 1997, 119, 2114–2118 CrossRef CAS.
- L.-X. Wang, Carbohydr. Res., 2008, 343, 1509–1522 CrossRef CAS.
- J. R. Rich and S. G. Withers, Nat. Chem. Biol., 2009, 5, 206–215 CrossRef CAS.
- F. Schwarz, W. Huang, C. Li, B. L. Schulz, C. Lizak, A. Palumbo, S. Numao, D. Neri, M. Aebi and L.-X. Wang, Nat. Chem. Biol., 2010, 6, 264–266 CrossRef CAS.
- K. Takegawa, S. Yamaguchi, A. Kondo, H. Iwamoto, M. Nakoshi, I. Kato and S. Iwahara, Biochem. Int., 1991, 24, 849–855 CAS.
- K. J. Yamamoto, S. Kadowaki, J. Watanabe and H. Kumagai, Biochem. Biophys. Res. Commun., 1994, 203, 244–252 CrossRef CAS.
- K. Fujita and K. Yamamoto, Biochim. Biophys. Acta, 2006, 1760, 1631–1635 CrossRef CAS.
- K. Takegawa, M. Tabuchi, S. Yamaguchi, A. Kondo, I. Kato and S. Iwahara, J. Biol. Chem., 1995, 270, 3094–3099 CrossRef CAS.
- B. Li, H. Song, S. Hauser and L.-X. Wang, Org. Lett., 2006, 8, 3081–3084 CrossRef CAS.
- C. D. Heidecke, Z. Ling, N. C. Bruce, J. W. B. Moir, T. B. Parsons and A. J. Fairbanks, ChemBioChem, 2008, 9, 2045–2051 CrossRef CAS.
- T. W. D. F. Rising, C. D. Heidecke, J. W. B. Moir, Z. Ling and A. J. Fairbanks, Chem.–Eur. J., 2008, 14, 6444–6464 CrossRef CAS.
- H. Ochiai, W. Huang and L.-X. Wang, J. Am. Chem. Soc., 2008, 130, 13790–13803 CrossRef CAS.
- W. Huang, H. Ochiai, X. Zhang and L.-X. Wang, Carbohydr. Res., 2008, 343, 2903–2913 CrossRef CAS.
- W. Huang, C. Li, B. Li, M. Umekawa, K. Yamamoto, X. Zhang and L.-X. Wang, J. Am. Chem. Soc., 2009, 131, 2214–2223 CrossRef CAS.
- Sequential chemoenzymatic methods have been successfully applied to glycoprotein production using first chemical attachment then use of glycosyltransferases or glycosynthases. These do not allow an en bloc approach and are limited to single sugar-by-sugar attachment with inherent potential compounding of sequential yields and substrate intolerance; see: H. Liu, L. Wang, A. Brock, C. H. Wong and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 1702–1703 Search PubMed; D. P. Gamblin, P. Garnier, S. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 43, 828–833 CrossRef CAS; J. M. Müllegger, H. M. Chen, R. A. J. Warren and S. G. Withers, Angew. Chem., Int. Ed., 2006, 45, 2585–2588 CrossRef CAS.
- L.-X. Wang, M. Tang, T. Suzuki, K. Kitajima, Y. Inoue, S. Inoue, J.-Q. Fan and Y. C. Lee, J. Am. Chem. Soc., 1997, 119, 11137–11146 CrossRef CAS.
- W. Huang, S. Groothuys, A. Heredia, B. H. M. Kuijpers, F. P. J. T. Rutjes, F. L.v. Delft and L.-X. Wang, ChemBioChem, 2009, 10, 1234–1242 CrossRef CAS.
- B. G. Davis, Pure Appl. Chem., 2009, 81, 285–298 CrossRef CAS.
- S. I. van Kasteren, H. B. Kramer, H. H. Jensen, S. J. Campbell, J. Kirkpatrick, N. J. Oldham, D. C. Anthony and B. G. Davis, Nature, 2007, 446, 1105–1109 CrossRef.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS.
- O. Boutureira, G. J. L. Bernardes, M. Fernández-González, J. M. Chalker and B. G. Davis, manuscript in preparation.
- D. P. Gamblin, P. Garnier, S. van Kasteren, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Angew. Chem., Int. Ed., 2004, 43, 828–833 CrossRef CAS.
- Y. Zeng, J. Wang, B. Li, S. Hauser, H. Li and L.-X. Wang, Chem.–Eur. J., 2006, 12, 3355–3364 CrossRef CAS.
- K. J. Doores, Y. Mimura, R. A. Dwek, P. M. Rudd, T. Elliott and B. G. Davis, Chem. Commun., 2006, 1401–1403 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- E. Akaike, M. Tsutsumida, K. Osumi, M. Fujita, T. Yamanoi, K. Yamamoto and K. Fujita, Carbohydr. Res., 2004, 339, 719–722 CrossRef.
- All prior Gal-containing acceptors (see above Ref) have proven inactive with Endo. Our results are not only consistent with these but also further confirm that the anticipated OH-4 regioselectivity of glycosylation is maintained on glycoprotein acceptors.
- O. Boutureira, F. D'Hooge, M. Fernández-González, Gonçalo J. L. Bernardes, M. Sánchez-Navarro, J. R. Koeppe and B. G. Davis, Chem. Commun., 2010 10.1039/c1030cc01576h.
- D. P. Gamblin, P. Garnier, S. J. Ward, N. J. Oldham, A. J. Fairbanks and B. G. Davis, Org. Biomol. Chem., 2003, 1, 3642–3644 RSC.
- O. Prante, J. R. Einsiedel, R. Haubner, P. Gmeiner, H.-J. r. Wester, T. Kuwert and S. Maschauer, Bioconjugate Chem., 2006, 18, 254–262.
- M. W. Vetting, S. S. Hegd, K. Z. Hazleton and J. S. Blanchard, Protein Sci., 2007, 16, 755 CrossRef CAS.
- K. Pachamuthu and R. R. Schmidt, Chem. Rev., 2006, 106, 160–187 CrossRef.
- B. Capon, Chem. Rev., 1969, 69, 407–498 CrossRef CAS.
- Z. J. Witczak, R. Chhabra, H. Chen and X.-Q. Xie, Carbohydr. Res., 1997, 301, 167–175 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, including 1H and 13C NMR spectra for all new compounds and ESI-MS for all protein samples. See DOI: 10.1039/c0sc00265h |
| This journal is © The Royal Society of Chemistry 2010 |