N-heterocyclic carbenes which readily add ammonia, carbon monoxide and other small molecules†,‡
Ulrich
Siemeling
*a,
Christian
Färber
a,
Clemens
Bruhn
a,
Michael
Leibold
a,
Detlef
Selent
b,
Wolfgang
Baumann
b,
Moritz
von Hopffgarten
c,
Catharina
Goedecke
c and
Gernot
Frenking
*c
aInstitut für Chemie, Universität Kassel, Heinrich-Plett-Str. 40, 34132, Kassel, Germany. E-mail: siemeling@uni-kassel.de; Fax: +49-561-804-4777; Tel: +49-561-804-4576
bLeibniz Institute for Catalysis e. V. at the University of Rostock, Albert-Einstein-Str. 29a, 18059, Rostock, Germany
cFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Str., 35032, Marburg, Germany. E-mail: frenking@chemie.uni-marburg.de; Fax: +49-6421-28-25566; Tel: +49-6421-28-25563
First published on 22nd October 2010
Abstract
N-Heterocyclic carbenes (NHCs) are extremely valuable as nucleophilic organocatalysts. They are widely applied as ligands in transition-metal catalysed reactions, where they are known as particularly potent σ-donors. They are commonly viewed as workhorses exhibiting reliable, but undramatic, chemical behaviour. The N → Ccarbene π-donation stabilises NHCs at the expense of low reactivity towards nucleophiles. In contrast to NHCs, stable (alkyl)(amino)carbenes exhibit spectacular reactivity, allowing, for example, the splitting of hydrogen and ammonia and the fixation of carbon monoxide. NHCs have been judged to be electronically not suitable for showing similar reactivity. Here, we demonstrate that a ferrocene-based NHC is able to add ammonia, methyl acrylate, tert-butyl isocyanide, and carbon monoxide—reactions typical of (alkyl)(amino)carbenes, but unprecedented for diaminocarbenes. We also show that even the simplest stable diaminocarbene, C(NiPr2)2, adds CO. This reaction affords a β-lactam by a subsequent intramolecular process involving a C–H activation. Our results shed new light on the chemistry of diaminocarbenes and offer great potential for synthetic chemistry and catalysis.
Introduction
Transient carbenes like CCl2 have been playing a prominent role as reactive intermediates in organic synthesis for more than half a century.1 The general belief that carbenes are always transient species was shattered by the synthesis of a stable, crystalline N-heterocyclic carbene (NHC) by Arduengo in 1991.2 A stable (phosphino)(silyl)carbene had already been prepared before by Bertrand, but it took several years before it was fully recognised as a carbon(II) compound.3 Since then, the chemistry of these and related heteroatom-stabilised singlet carbenes has experienced tremendous development.4–8Singlet carbenes can exhibit electrophilic and nucleophilic properties due to the presence of a vacant p-type orbital (LUMO) and an occupied orbital perpendicular to this empty orbital which contains the lone pair of electrons at the divalent carbon atom (HOMO). The nature of the substituents X and Y has a decisive influence on the degree of electrophilicity and nucleophilicity of the carbene X–C–Y.9 For example, the transient CCl2 exhibits pronounced electrophilicity9–13 and only latent nucleophilicity.14 Stable singlet carbenes have been classified in three categories.4,15 Push–pull carbenes contain a π-donor substituent which interacts with the vacant p-type orbital and a π-acceptor substituent which interacts with the orbital occupied by the lone pair. This results in a quasilinear polarised heterocumulene-type structure. The most representative carbenes of this type are (phosphino)(silyl)carbenes,16,17 which show noticeable electrophilic behaviour.18 Push–spectator carbenes form the second category. They contain only a single electron-active substituent, which is π-donating. Typical examples of this kind are (alkyl)(amino)carbenes,16 which are extremely reactive and even able to add fundamentally important small molecules like H2, NH3, and CO.19,20 By far the largest category are push–push carbenes, whose substituents X and Y are both π-donating. This results in a four-electron three-centre π-system and a particularly low degree of electrophilicity. The iconic representatives are NHCs. Carbenes which belong to the latter two categories are bent.
NHCs, especially those derived from the five-membered heterocycles imidazole and imidazoline, are the most widely investigated subclass of singlet carbenes. Imidazole-based NHCs are thermodynamically stable towards dimerisation to the corresponding enetetramines. Imidazoline-based NHCs and their non-cyclic analogues of the type C(NR2)221 can be made kinetically inert towards this reaction by bulky N-substituents. NHCs have proved to be extremely useful as organocatalysts22 and as ligands in transition-metal catalysed reactions, where they can act as particularly potent σ-donors, stronger even than the electron-rich trialkylphosphanes.23,24 These applications are based on the high σ-donicity and nucleophilicity of diaminocarbenes.
In contrast to (alkyl)(amino)carbenes,19,20 which belong to the push–spectator category, diaminocarbenes have been judged to be not sufficiently electrophilic to add small molecules like H2 and CO.25 It is a challenge to achieve such reactions with diaminocarbenes. A strategy to increase their electrophilicity by shepherding nitrogen lone-pair donation away from the empty p-type orbital at the divalent carbon atom was recently reported.25,26 This effect was achieved by replacing the diamino by a diamido structure in a six-membered ring. The cyclic diamidocarbenes 1-DIPP (R = 2,6-diisopropylphenyl) and 1-Mes (R = mesityl) turned out to be sufficiently electrophilic to add isocyanides, affording diamidoketenimines 2 (Scheme 1).27,28 Such coupling reactions are typical of transient carbenes like CCl2.29 The only stable singlet carbenes which had previously shown this behaviour are push–pull type (phosphino)(silyl)carbenes17 and (phosphino)(aryl)carbenes30 as well as push–spectator type (amino)(aryl)carbenes.31 Even the addition of CO proved possible with 1-DIPP and 1-Mes, affording the ketene derivatives of type 3 in a reversible reaction of low thermodynamic driving force (Keq ≤ 5 M−1 at room temperature) (Scheme 1).25,28 An analogous carbonylation had previously been reported only for push–spectator type (alkyl)(amino)carbenes, whose CO binding is, however, considerably stronger.20 Even more spectacular, these (alkyl)(amino)carbenes were shown to add NH3 in a concerted reaction best described as a nucleophilic activation of ammonia.19 NH3 also adds to 1-DIPP and 1-Mes, respectively, under mild conditions, affording products of type 4 (Scheme 1).27 A nucleophilic activation of ammonia seems very unlikely here.
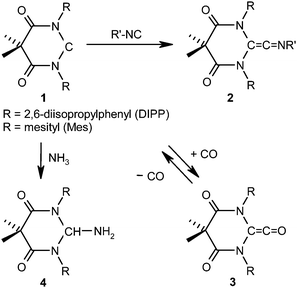 | ||
| Scheme 1 Comparatively electrophilic diamidocarbenes of type 1. Addition reactions of 1-DIPP (R = 2,6-diisopropylphenyl) and 1-Mes (R = mesityl) with isocyanides, carbon monoxide, and ammonia afford ketenimines 2, ketenes 3, and amino derivatives 4, respectively. | ||
The replacement of the flanking electron-donating amino groups by electron-withdrawing amido units leads to an increase of the carbene's electrophilicity at the expense of its σ-donicity. The σ-donor strength of singlet carbene ligands is commonly judged by the Tolman electronic parameter (TEP), which can be derived from IR spectroscopic data of carbonyl complexes.32,33 The TEP values of 2057 cm−1 and 2055 cm−1 for 1-DIPP and 1-Mes,26 respectively, show that such diamidocarbenes are much weaker σ-donors than cyclic diaminocarbenes with a six-membered ring system (TEP ≤ 2045 cm−1) and even weaker than standard imidazole- and imidazoline-based NHCs, whose TEP values as a rule are below ca. 2052 cm−1. These TEP values were calculated from experimental data25,26,34 using the linear regressions described in ref. 32 and 33
We are following an alternative approach to modulate the electronic properties of NHCs by oxidation of a redox-active carbene backbone and recently reported the first examples of stable, crystalline [3]ferrocenophane-type NHCs 5 (Fig. 1), where formally a d-block metal atom constitutes an integral part of a six-membered ring.34,35 Independently from us, analogues with less bulky N-substituents that consequently were too unstable for isolation were described by others.36 NHCs of type 5 exhibit TEP values of ca. 2045 cm−1, in accord with a σ-donor strength much higher than that of traditional imidazole- and imidazoline-based NHCs. The stable neopentyl and 2-adamantyl derivatives 5-Np and 5-Ad can be easily oxidised to the corresponding delocalised radical cations, whose persistent nature is unprecedented in NHC chemistry.34
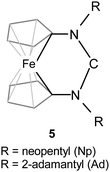 | ||
| Fig. 1 Ferrocene-based N-heterocyclic carbenes of type 5. The structure is drawn in a way that highlights the six-membered heterocyclic ring. | ||
Results and discussion
We have focused our study on compound 5-Np, since the solubility and crystallinity of this carbene and its complexes turned out to be superior to that of its 2-adamantyl analogue 5-Ad. During the course of our investigation, we noticed that this NHC exhibits a reactivity similar to that reported for the push–spectator type (alkyl)(amino)carbenes. This was hitherto thought to be impossible for diaminocarbenes. The relevant reactions of 5-Np are summarised in Scheme 2.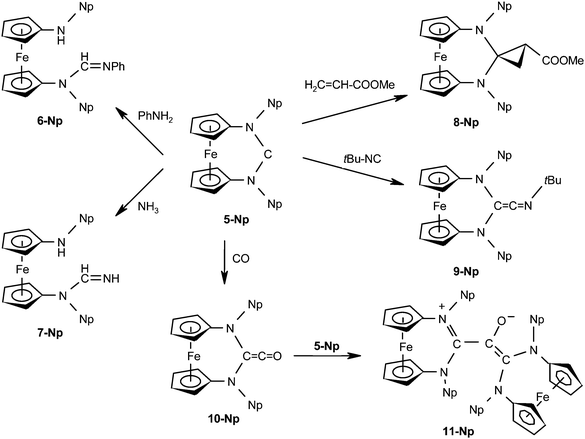 | ||
| Scheme 2 Addition reactions of 5-Np with aniline, ammonia, methyl acrylate, tert-butyl isocyanide, and carbon monoxide, respectively affording the amino-amidine derivatives 6-Np and 7-Np, the cyclopropane derivative 8-Np, the ketenimine derivative 9-Np, and the zwitterionic compound 11-Np. | ||
5-Np adds aniline, forming the amino-amidine 6-Np. Analogous products had been obtained before in reactions of enetetramines (R2N)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(NR2)2 with primary amines. There is no evidence for the participation of transient diaminocarbenes C(NR2)2 in this process.37 In the same vein, 5-Np affords the open-chain addition product 7-Np with ammonia. The addition of NH3 to a stable singlet carbene had previously been reported only for diamidocarbenes of type 127 and push–spectator type (alkyl)(amino)carbenes.196-Np and 7-Np were obtained as oils which showed no tendency to crystallise, as is typical for this class of compounds.38
C(NR2)2 with primary amines. There is no evidence for the participation of transient diaminocarbenes C(NR2)2 in this process.37 In the same vein, 5-Np affords the open-chain addition product 7-Np with ammonia. The addition of NH3 to a stable singlet carbene had previously been reported only for diamidocarbenes of type 127 and push–spectator type (alkyl)(amino)carbenes.196-Np and 7-Np were obtained as oils which showed no tendency to crystallise, as is typical for this class of compounds.38
5-Np reacts with the electron-poor olefin methyl acrylate, affording the cyclopropane derivative 8-Np. Such reactions had previously been reported for push–pull type (phosphino)(silyl)carbenes39 and for a push–spectator type (alkyl)(amino)carbene,40 but not for diaminocarbenes. 8-Np was structurally characterised by a single-crystal X-ray diffraction study (Fig. 2). We note that the nitrogen atoms are pyramidalised (sum of angles ca. 346° and 349° at N1 and N2, respectively) and the average N–C1 bond length of ca. 1.46 Å is considerably longer than that in the carbene 5-Np (ca. 1.36 Å), which is in accord with a loss of π-delocalisation in the N–C–N unit upon cyclopropane formation.
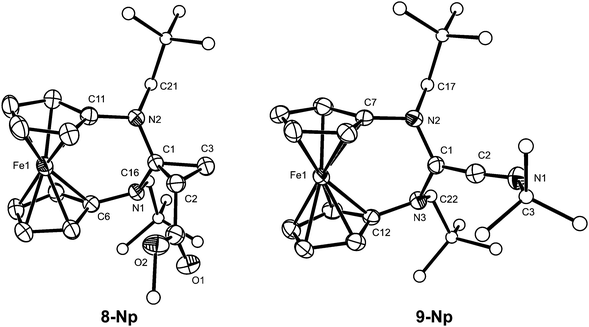 | ||
| Fig. 2 Molecular structures of 8-Np and 9-Np in the crystal. ORTEP plots with thermal ellipsoids drawn at the 30% probability level. For clarity, H atoms are omitted and the C atoms of the neopentyl substituents, the OMe group of 8-Np and the tert-butyl group of 9-Np are displayed as circles of arbitrary size. Selected bond lengths (Å) and angles (°) for 8-Np: C1–N1 1.454(4), C1–N2 1.456(4), C1–C2 1.534(4), C1–C3 1.491(4), C2–C3 1.517(5), C6–N1 1.429(4), C11–N2 1.436(4), N1–C1–N2 119.6(2), C6–N1–C1 116.0(2), C6–N1–C16 115.4(2), C1–N1–C16 114.3(2), C1–N2–C11 116.3(2), C1–N2–C21 116.6(2), C11–N2–C21 116.6(2); for 9-Np: C1–C2 1.318(5), C1–N2 1.435(4), C1–N3 1.439(4), C2–N1 1.232(5), C3–N1 1.490(5), C7–N2 1.433(4), C12–N3 1.439(4), N2–C1–N3 121.9(3), N1–C2–C1 172.3(4), C2–N1–C3 122.4(3), C1–N2–C7 115.1(3), C1–N2–C17 118.2(3), C7–N2–C17 117.9(3), C1–N3–C12 114.5(3), C1–N3–C22 117.8(3), C12–N3–C22 117.4(3). | ||
5-Np reacts smoothly and swiftly with tert-butyl isocyanide, affording the diaminoketenimine 9-Np, which was structurally characterised by a single-crystal X-ray diffraction study (Fig. 2). The C1–C2–N1 unit is quasilinear and the bond lengths in this unit (C1–C2 1.318(5) Å, C2–N1 1.232(5) Å) are in accord with substantial multiple bond character, in line with data reported for a number of closely related compounds of type 2.28 The isocyanide addition causes a pronounced elongation of the average N–C bond length in the N–C–N unit of the carbene 5-Np from ca. 1.36 Å to ca. 1.44 Å and a pyramidalisation of the nitrogen atoms in this unit (sum of angles ca. 351° and 350° at N2 and N3, respectively), which is similar to the observations described above for 8-Np.
5-Np also adds carbon monoxide. This reaction obviously affords the diaminoketene 10-Np, which is efficiently trapped by nucleophilic addition of 5-Np to afford the final zwitterionic product 11-Np, whose structure was elucidated by a single-crystal X-ray diffraction study (Fig. 3). Similar zwitterionic products had previously been obtained in reactions of enetetramines (R2N)2CC(NR2)2 with diphenylketene.37 In agreement with data reported for other enolate zwitterions,41 the C1–C2 and C2–O1 bonds of the enolate core (1.384(9) Å and 1.301(8) Å, respectively) both possess partial double bond character, while the C2–C3 distance of 1.534(10) Å corresponds to a single bond. A geometry optimisation of 11-Np where the C1–C2 and C2–C3 bonds were constrained to have the same length gave a C2-symmetric structure which is 6.9 kcal mol−1 higher in energy at BP86/def2-TZVPP//BP86/def2-SVP than the equilibrium form. The N-heterocyclic unit containing C3 exhibits bond parameters compatible with a π-delocalised formamidinium-type structure (average C3–N bond length ca. 1.36 Å) with essentially trigonal planar N atoms (sum of angles 359.2° and 357.2°), while the unit containing C1 exhibits pyramidalised N atoms (sum of angles 344.4° and 348.7°) and considerably longer carbon–nitrogen bonds (average C1–N ca. 1.45 Å), similar to the corresponding parameters observed for 8-Np and 9-Np.
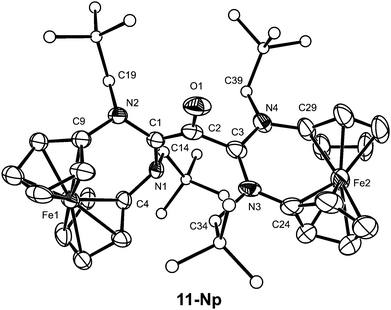 | ||
| Fig. 3 Molecular structure of 11-Np in the crystal. ORTEP plot with thermal ellipsoids drawn at the 30% probability level. For clarity, H atoms are omitted and the C atoms of the neopentyl substituents are displayed as circles of arbitrary size. Selected bond lengths (Å) and angles (°): C1–C2 1.384(9), C1–N1 1.463(7), C1–N2 1.436(8), C2–O1 1.301(8), C2–C3 1.534(10), C3–N3 1.361(8), C3–N4 1.362(9), C4–N1 1.449(7), C9–N2 1.420(8), C24–N3 1.404(11), C29–N4 1.446(11), N1–C1–N2 121.2(5), O1–C2–C1 129.3(7), O1–C2–C3 107.0(6), C1–C2–C3 123.6(6), N3–C3–N4 123.7(6), C1–N1–C4 115.6(4), C1–N1–C14 116.1(5), C4–N1–C14 112.7(5), C1–N2–C9 115.7(5), C1–N2–C19 116.1(4), C9–N2–C19 116.9(5), C3–N3–C24 126.9(6), C3–N3–C34 118.3(6), C24–N3–C34 114.0(5), C3–N4–C29 126.3(6), C3–N4–C39 123.3(6), C29–N4–C39 107.6(6). | ||
Such a follow-up reaction was not reported for the ketene derivatives obtained with (alkyl)(amino)carbenes20 and with the diamidocarbenes of type 1.25 This difference in behaviour is probably due to several factors. Substituents with π-donor and σ-acceptor character like NR2 destabilise ketenes.42 Aminoketenes are therefore highly reactive, transient species. While less electron-rich amido-43 and phthalimido-derived ketenes44 had already been isolated decades ago, it was only recently that the first examples of stable aminoketenes were described. They contain a single, very bulky amino group.45 Owing to the presence of two amino substituents, 10-Np is expected to be extremely reactive, certainly much more than a diamidoketene of type 3. It is therefore rapidly attacked by the NHC 5-Np, which is a particularly potent nucleophile in comparison to 1-DIPP and 1-Mes, as indicated by the corresponding TEP values (vide supra). In the case of the aminoketenes obtained with very bulky (alkyl)(amino) carbenes, the follow-up reaction is unlikely for steric reasons.
The reactivity of the diaminocarbene 5-Np towards ammonia, methyl acrylate, tert-butyl isocyanide, and carbon monoxide is strongly reminiscent of the behaviour of push–spectator type (alkyl)(amino)carbenes. According to quantum-chemical calculations, these are slightly more nucleophilic, but considerably more electrophilic, than traditional NHCs.19,46 We have performed DFT calculations to gain further insight into the reactivity of NHCs of type 5 (Table 1). For comparison, we have also analysed the behaviour of several other related carbenes, namely the diamidocarbene 1-Me, the (alkyl)(amino)carbene 12-Me, the acyclic diaminocarbene C(NMe2)2 (13-Me), and 1,3-dimethylimidazol-2-ylidene (14-Me) towards CO. The N-substituent R was chosen as methyl in these model compounds. Furthermore, the stable isopropyl-substituted acyclic diaminocarbene 13-iPr was also included in our study. Compound 12-Me is a simplified model for the cyclic (alkyl)(amino)carbene 12′-DIPP, which is known to add CO.20 The only previous closely related quantum-chemical study confirmed that imidazole-based NHCs do not react with carbon monoxide.47Table 1 shows the calculated activation barriers and reaction energies for the addition reactions of the carbenes with CO. The data predict that the addition of CO to all carbenes which have been calculated takes places with rather low activation barriers. The calculations also suggest that the CO addition to 14-Me is endoenergetic and endergonic at 298.15 K and 1 atm, while the reactions of the other carbenes with carbon monoxide are exoenergetic and exergonic. This is in agreement with experimental observations.20,25,47 The former reaction yields a weakly bonded complex between CO and 14-Me where the CO is bound to the carbene centre with a C–C–O angle of 122.2° and with a long C–C distance of 1.626 Å (Fig. S1, Supporting Information‡). This complex is only a very shallow local energy minimum on the potential energy surface which, after zero-point energy contributions are considered, is even 0.2 kcal mol−1 higher in energy than the transition state. This finding has been reported before.47 All other investigated carbenes react with CO yielding a ketene, as indicated by essentially linear coordination of CO and short C–C distances between the carbon monoxide ligand and the carbene centre (see Table S1, Supporting Information‡).
| ΔE2/kcal mol−1 | ΔG2/kcal mol−1 | ΔER/kcal mol−1 | ΔGR/kcal mol−1 | ΔES–T/kcal mol−1 | E HOMO/eV | |
|---|---|---|---|---|---|---|
| 1-Me | 1.4 | 3.8 | −19.8 | −16.1 | 42.2 | −4.96 |
| 5-Me | 2.8 | 5.2 | −15.5 | −11.8 | 36.9 | −4.03 |
| 12-Me | 1.7 | 3.6 | −28.2 | −25.2 | 46.8 | −4.02 |
| 13-Me | 2.7 | 4.7 | −17.3 | −14.9 | 37.8 | −3.69 |
| 13-iPr | 7.3 | 13.6 | −17.2 | −12.1 | 41.0 | −3.50 |
| 14-Me | 6.5 | 10.1 | 6.7 | 9.9 | 83.3 | −4.51 |
We calculated the complete reaction path for the addition of CO to 5-Me yielding the intermediate ketene 10-Me and the subsequent nucleophilic attack of another molecule 5-Me, which leads to the final product 11-Me. The results are presented in Fig. 4. The calculated data suggest that the ketene 10-Me, which is formed by addition of CO to 5-Me with a low activation barrier (ΔG‡ = 5.2 kcal mol−1), reacts easily with another carbene 5-Me in a second step yielding the final product 11-Me, which has an even lower activation barrier with respect to a precoordinated complex (ΔG‡ = 3.9 kcal mol−1) in a highly exothermic and exergonic reaction (ΔGR = −21.1 kcal mol−1). This explains why the ketene 10-Me could not be isolated as a reaction product. The calculated bond lengths of the final reaction product 11-Me are in good agreement with experimental data of 11-Np in the crystal. The nitrogen atoms in the ketene 10-Me exhibit strong pyramidalisation and the CO moiety is bound almost linearly (177.5°) to the former carbene centre with a distance of 1.346 Å. The C–O bond in the adduct is slightly elongated (1.179 Å) with respect to free carbon monoxide (1.142 Å).
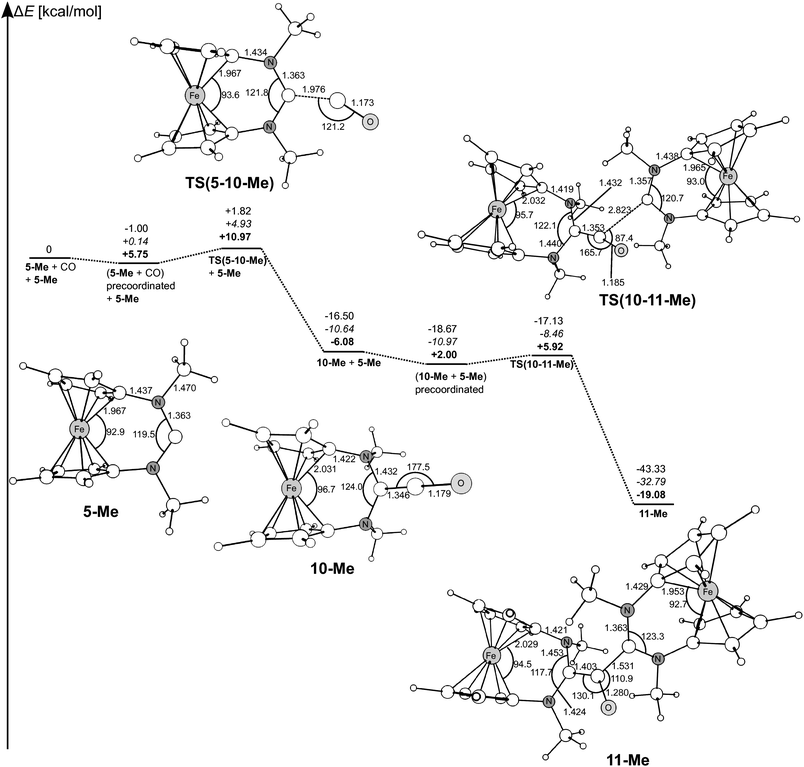 | ||
| Fig. 4 Calculated reaction pathway for the reaction of 5-Me with CO to ketene 10-Me and subsequent nucleophilic attack. Relative energies with respect to the free reactants. Numbers in normal font give relative energies ΔEo calculated at BP86/def2-SVP. Numbers in italics give ΔEo at BP86/def2-TZVPP//BP86/def2-SVP. Numbers in boldface give relative Gibb's Free Energies ΔG at 298.15 K and 1 atm at BP86/def2-SVP. Distances are given in Å, angles in degrees. | ||
In the second reaction step, 10-Me + 5-Me → 11-Me, carbene 5-Me reacts as a nucleophile with ketene 10-Me. Such a reaction was not observed for the diamidocarbenes 1-Mes and 1-DIPP in their reactions with CO.25 The nucleophilicity of a singlet carbene is indicated by the energy of its HOMO (EHOMO). Table 1 shows that the HOMO energy of 5-Me (−4.03 eV) is quite similar to that of the (alkyl)(amino)carbene 12-Me (−4.02 eV) and the acyclic diaminocarbene 13-Me (−3.69 eV), but significantly higher than that of the diamidocarbene 1-Me (−4.96 eV). The stable acyclic diaminocarbene 13-iPr exhibits the highest HOMO energy (−3.50 eV) in this series. It has been suggested that the electrophilicity of a singlet carbene is reliably indicated by its singlet–triplet gap ΔES–T.48Table 1 shows that the imidazole-based NHC 14-Me has a much higher singlet–triplet gap of 83.3 kcal mol−1 than the other carbenes, which have rather similar ΔES–T values between 36.9 and 46.8 kcal mol−1 and are therefore considerably more electrophilic than traditional NHCs.
These computational results led us to the conclusion that, like ferrocene-based diaminocarbenes of type 5, but in contrast to traditional NHCs, stable acyclic diaminocarbenes like C(NiPr2)2 (13-iPr)21 should also be able to react with CO. Indeed, this turned out to be the case. Despite the particularly high HOMO energy (and hence nucleophilicity) of 13-iPr, a follow-up reaction of the diaminoketene (iPr2N)2CCO (15-iPr) with 13-iPr to a zwitterionic product analogous to 11-Np was not observed, which is most likely due to steric reasons. Instead, 15-iPr obviously undergoes an intramolecular process which involves the activation of a C–H bond and results in the formation of the azetidin-2-one (β-lactam) derivative 16-iPr (Scheme 3). The product was isolated as a light yellow oil which showed no tendency to crystallise. Its chemical composition was established by high-resolution mass spectrometry. The azetidin-2-one structure is evident already from the diagnostic ν(CO) band located at 1741 cm−1 in the IR spectrum and the resonance signal observed for the carbonyl C atom at δ = 167 ppm in the 13C NMR spectrum. All pertinent spectroscopic data collected for 16-iPr agree very well with those reported for the benzyl analogue 16-Bz (RCH2Ph).49
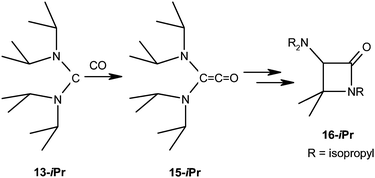 | ||
| Scheme 3 Addition reaction of the stable acyclic diaminocarbene 13-iPr with carbon monoxide, affording the β-lactam 16-iPr. | ||
The β-lactams are one of the best known and intensively investigated ring systems due to both their utility as synthetic intermediates and their biological activity as antibiotics, and new synthetic methods for the generation of this prominent class of compounds are consequently of great interest.50,51 The formation of 16-iPr is an example of a new synthetic entry to this important ring system. The reaction of 13-iPr with CO was carefully monitored in situ by NMR spectroscopy. No reaction was observed in toluene solution below ca. −20 °C. Above that temperature, 16-iPr was the only detectable species present apart from unreacted 13-iPr and CO, which reflects the high reactivity of the transient diaminoketene (iPr2N)2CCO (15-iPr). It is tempting to surmise that the initial step of the intramolecular process following ketene formation is a nucleophilic attack of a diisopropylamino nitrogen atom at the central carbon atom of the ketene moiety. We calculated a possible pathway for this reaction. The results are shown in Fig. 5. The nucleophilic attack of a nitrogen atom on the ketene moiety proceeds with a moderate activation barrier (ΔG‡ = 17.5 kcal mol−1), which is compatible with our finding that the reaction was not observed at low temperatures. This step results in the formation of the intermediate carbene iPr2N–C–C(O)NiPr2 (17), which contains a π-electron donating amino substituent together with a π-electron withdrawing carboxamido substituent. The subsequent insertion of this push–pull carbene into the Me2C–H bond of one of the isopropyl substituents proceeds with an activation barrier of 8.0 kcal mol−1. Note that the carbene character of the first stable push–pull carbene was inferred from its insertion into an unpolarised C–H bond.3,52 The formation of the β-lactam 16-iPr from the ketene (iPr2N)2CCO (15-iPr) is more than twice as exergonic (ΔGR = −26.4 kcal mol−1) as the formation of this ketene from the acyclic diaminocarbene 13-iPr (−12.1 kcal mol−1, Table 1). A study which addresses the scope of this novel reaction by utilising a range of other stable or persistent acyclic diaminocarbenes5 is currently underway.
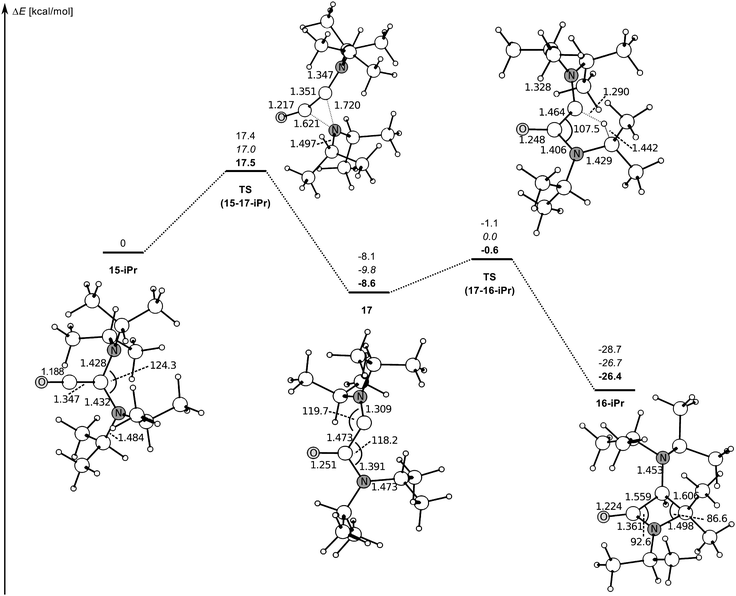 | ||
| Fig. 5 Calculated reaction pathway for the reaction of ketene 15-iPr to β-lactam 16-iPr (relative energies in kcal mol−1 with respect to the ketene). Numbers in normal font give energies ΔEo including zero-point correction at BP86/def2-SVP. Numbers in italics give ΔEo at BP86/def2-TZVPP//BP86/def2-SVP using the zero-point correction calculated at BP86/def2-SVP. Numbers in boldface give Gibbs' Free Energies ΔG at 298.15 K and 1 atm at BP86/def2-SVP. Distances are given in Å, angles in degrees. | ||
Conclusions
The results which are presented here demonstrate that, contrary to previous beliefs, stable NHCs can show strongly enhanced reactivities towards fundamentally important nucleophiles like ammonia, isocyanides, and carbon monoxide, which had previously been observed only for (alkyl)(amino)carbenes and the particularly electrophilic diamidocarbenes. The ferrocene-based NHC system investigated in our study is electronically closely related to (alkyl)(amino)carbenes, exhibiting both a high degree of nucleophilicity (indicated by a high HOMO energy) and a high degree of electrophilicity (indicated by a small singlet–triplet gap). The hitherto unrecognised reactivity of the simplest stable diaminocarbene, viz. Alder's C(NiPr2)2, towards carbon monoxide, which results in a novel synthetic entry to β-lactams, further emphasises that diaminocarbenes have been much underestimated in their synthetic potential. Our observations open the door to an unexplored and exciting area of synthetically useful carbene chemistry.Acknowledgements
We are grateful to the Deutsche Forschungsgemeinschaft for generous financial support (SI 429/16-2) and to Professor Kees Elsevier (University of Amsterdam) for helpful suggestions.Notes and references
- M. B. Smith and J. March, March's Advanced Organic Chemistry, 6th ed., Wiley, Hoboken, NJ, 2007, pp. 283–292 Search PubMed.
- A. J. Arduengo, III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1989, 28, 621 CrossRef.
- T. Kato, E. Maerten and A. Baceiredo, in Transition Metal Complexes of Neutral η1-Carbon Ligands (Top. Organomet. Chem., 30), ed. R. Chauvin and Y. Canac, Springer, Berlin, 2010, pp. 131–147 Search PubMed.
- J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333 CrossRef CAS.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862 CrossRef CAS.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef.
- J.-L. Mieusset and U. H. Brinker, J. Org. Chem., 2008, 73, 1553 CrossRef CAS.
- J. Oláh, F. De Proft, T. Veszprémi and P. Geerlings, J. Phys. Chem. A, 2004, 108, 490 CrossRef CAS.
- P. Pérez, J. Phys. Chem. A, 2003, 107, 522 CrossRef CAS.
- W. Sander, C. Kötting and R. Hübert, J. Phys. Org. Chem., 2000, 13, 561 CrossRef CAS.
- R. A. Moss, Acc. Chem. Res., 1989, 22, 15 CrossRef CAS.
- R. A. Moss, M. Zhang and K. Krogh-Jespersen, Org. Lett., 2009, 11, 1947 CrossRef CAS.
- Y. Canac, S. Conejero, B. Donnadieu, W. W. Schoeller and G. Bertrand, J. Am. Chem. Soc., 2005, 127, 7312 CrossRef CAS.
- Y. Canac, M. Soleilhavoup, S. Conejero and G. Bertrand, J. Organomet. Chem., 2004, 689, 3857 CrossRef CAS.
- G. Bertrand and R. Reed, Coord. Chem. Rev., 1994, 137, 323 CrossRef CAS.
- S. Goumri-Magnet, O. Polishchuk, H. Gornitzka, C. J. Marsden, A. Baceiredo and G. Bertrand, Angew. Chem., Int. Ed., 1999, 38, 3727 CrossRef.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS.
- V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488 CrossRef CAS.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121 CrossRef.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS.
- F. Glorius, ed., N-Heterocyclic Carbenes in Transition Metal Catalysis (Top. Organomet. Chem., 21), Springer, Berlin, 2007 Search PubMed.
- S. P. Nolan, ed., N-Heterocyclic Carbenes in Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed.
- T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039 CrossRef CAS.
- V. César, N. Lugan and G. Lavigne, Eur. J. Inorg. Chem., 2010, 361 CrossRef CAS.
- T. W. Hudnall, J. P. Moerdyk and C. W. Bielawski, Chem. Commun., 2010, 46, 4288 RSC.
- T. W. Hudnall, E. J. Moorhead, D. G. Gusev and C. W. Bielawski, J. Org. Chem., 2010, 75, 2763 CrossRef CAS.
- A. Halleux, Angew. Chem., Int. Ed. Engl., 1964, 3, 752 CrossRef.
- E. Despagnet-Ayoub, H. Gornitzka, D. Bourissou and G. Bertrand, Eur. J. Org. Chem., 2003, 2039 CrossRef CAS.
- S. Solé, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2001, 292, 1901 CrossRef CAS.
- S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487 CrossRef CAS.
- R. A. Kelly III, H. Clavier, S. Guidice, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202 CrossRef.
- U. Siemeling, C. Färber, M. Leibold, C. Bruhn, P. Mücke, R. F. Winter, B. Sarkar, M. von Hopffgarten and G. Frenking, Eur. J. Inorg. Chem., 2009, 4607 CrossRef CAS.
- U. Siemeling, C. Färber and C. Bruhn, Chem. Commun., 2009, 98 RSC.
- D. M. Khramov, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Angew. Chem., Int. Ed., 2008, 47, 2267 CrossRef CAS.
- J. Hocker and R. Merten, Angew. Chem., Int. Ed. Engl., 1972, 11, 964 CAS.
- J. Hocker and R. Merten, Chem. Ber., 1972, 105, 1651 CrossRef CAS.
- S. Goumri-Magnet, T. Kato, H. Gornitzka, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 2000, 122, 4464 CrossRef CAS.
- V. Cavallo, J. Mafhouz, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 8670 CrossRef CAS.
- X.-F. Zhu, C. E. Henry and O. Kwon, J. Am. Chem. Soc., 2007, 129, 6722 CrossRef.
- L. Gong, M. A. McAllister and T. T. Tidwell, J. Am. Chem. Soc., 1991, 113, 6021 CrossRef CAS.
- P. Ganis, G. Paiaro, L. Pandolfo and G. Valle, Organometallics, 1988, 7, 210 CrossRef CAS.
- S. Winter and H. Pracejus, Chem. Ber., 1966, 99, 151 CrossRef CAS.
- T. T. Tidwell, Angew. Chem., Int. Ed., 2006, 45, 5580 CrossRef CAS.
- C.-L. Lai, W.-H. Guo, M.-T. Lee and C.-H. Hu, J. Organomet. Chem., 2005, 690, 5867 CrossRef CAS.
- D. A. Dixon, A. J. Arduengo, III, K. D. Dobbs and D. V. Khasnis, Tetrahedron Lett., 1995, 36, 645 CrossRef CAS.
- R. D. Bach, M.-D. Su, E. Aldabbagh, J. L. Andrés and H. B. Schlegel, J. Am. Chem. Soc., 1993, 115, 10237 CrossRef CAS.
- L. S. Hegedus and S. D'Andrea, J. Org. Chem., 1988, 53, 3113 CrossRef CAS.
- J. Marco-Contelles, Angew. Chem., Int. Ed., 2004, 43, 2198 CrossRef CAS.
- P. A. Magriotis, Angew. Chem., Int. Ed., 2001, 40, 4377 CrossRef CAS.
- A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Jürgen Heck on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental section, crystallographic data, computational details, supplementary table and figure. CCDC reference numbers 782899–782901. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00451k |
| This journal is © The Royal Society of Chemistry 2010 |