Continuous flow multi-step organic synthesis
Damien
Webb
* and
Timothy F.
Jamison
*
Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave., Cambridge, MA 02139, USA. E-mail: dwebb@mit.edu; tfj@mit.edu
First published on 23rd September 2010
Abstract
Using continuous flow techniques for multi-step synthesis enables multiple reaction steps to be combined into a single continuous operation. In this mini-review we discuss the current state of the art in this field and highlight recent progress and current challenges facing this emerging area.
 Damien Webb | Damien Webb was born and brought up in Barnsley, Yorkshire (England). He received an MChem degree from the University of Salford in 2005, during which time he spent a year working at the GSK Medicines Research Centre in Stevenage. Damien then obtained his Ph.D. from the University of Cambridge where he conducted natural product total synthesis research with Professor Steven V. Ley. In 2009 he joined the laboratories of Professor Timothy F. Jamison at MIT where his postdoctoral research is focused on the development of continuous methods for synthetic organic chemistry. |
 Timothy F. Jamison | Tim Jamison was born in San Jose, CA, and grew up in neighbouring Los Gatos, CA. He conducted undergraduate research with Professor Henry Rapoport at UC Berkeley for nearly three years and was then a Fulbright Scholar with Professor Steven A. Benner at the ETH Zürich. Thereafter he undertook his Ph.D. studies at Harvard University with Professor Stuart L. Schreiber and moved to the laboratory of Professor Eric N. Jacobsen at Harvard University, where he was a Damon Runyon-Walter Winchell postdoctoral fellow. In July 1999 he began his independent career at MIT, where he is currently Professor of Chemistry. He has been a principal investigator in the Novartis-MIT Center for Continuous Manufacturing since 2008. |
Introduction
The multi-step synthesis of complex organic compounds from simpler precursors is one of the outstanding accomplishments and ongoing challenges of synthetic organic chemistry. Through the development and invention of synthesis strategies, methods and technologies, increasingly complex molecules can be assembled with designed structures and functions for a variety of medicinal, agrochemical and materials applications. However, despite significant advances, organic synthesis is still considered highly labour- and resource-intensive.1The traditional pathway for multi-step synthesis proceeds by the batchwise and iterative step-by-step transformation of starting materials into desired products (Fig. 1(a)). Typically, after the completion of each synthetic step (A + B → C, C → D and D → E), products are isolated from the reaction mixture and purified to remove any undesired components that might interfere with the subsequent synthetic transformations. Although this approach is the foundation on which modern synthesis has been built, such an approach is time-consuming, often wasteful and in stark contrast to the single-cell multi-step biosynthetic pathways found in nature.2
 | ||
| Fig. 1 Multi-step synthesis strategies. | ||
A number of innovative strategies have been developed to increase synthetic efficiency.3 An increasingly popular approach to streamlining multi-step syntheses is the use of continuous flow techniques4 to combine multiple synthetic steps into a single, continuous and uninterrupted reactor network, thereby circumventing the need to isolate intermediate products (Fig. 1(b)). Such an approach has been called a ‘one-flow, multi-step synthesis’.5 The use of continuous flow methods for single-step transformations has been extensively reviewed elsewhere and will not be discussed in this mini-review. Here we detail some recent developments in the field of multi-step continuous flow synthesis6 and discuss and comment upon select contemporary examples of this emerging technology.
Multi-step flow synthesis
Solution-based approaches
The associated benefits of microreactors and continuous flow have been well documented.4 Typically, excellent mixing and temperature control is observed meaning the technique is ideally suited for performing extremely fast and exothermic reactions. The Yoshida group has published several examples outlining the use of highly reactive and unstable organolithium compounds for multi-step synthesis under continuous flow conditions.7 For example, o-dibromobenzene could be effectively coupled with two different electrophiles via sequential halogen–lithium exchange reactions in an extremely fast yet controlled manner (Scheme 1).8 The authors used flow reactors constructed from stainless steel micromixers and tubes, whilst the reagent streams were driven by syringe pump devices. The success of these protocols is attributed to effective temperature and residence time9 (tR) control that allows the unstable intermediates to be rapidly transferred to the next stage of the reactor before decomposition can occur. | ||
| Scheme 1 Generation and reaction of o-bromophenyllithium species using flow chemistry (Yoshida8). | ||
Importantly, the ability to scale-up this technique has also been demonstrated.10 Using the commercially available CYTOS microreactor system,11 a two-stage system was used for the lithiation and subsequent formylation of 3-bromo-anisole (Scheme 2).
 | ||
| Scheme 2 Generation of kilogram quantities of 3-methoxybenzaldehyde using flow chemistry (Schwalbe11). | ||
This reaction had proven difficult to reproduce on kilogram scale using batch methods (24% yield, −40 °C) owing to the long reagent dosing times that were required for effective temperature control. The efficient heat transfer properties of the microreactor system allowed the reaction to be conducted at a higher temperature (0 °C) and with short residence times (0.19 and 0.15 min), providing the desired product in 88% yield. A 24 h experiment afforded 1.4 kg of product at a throughput of 59 g h−1.
Workers at Bristol-Myers Squibb recently described the development of a pilot plant process for the continuous enolisation and oxidation of buspirone (Scheme 3).12 Again, the use of a microreactor system allowed the reactions to be conducted at elevated temperatures compared to the batch synthesis (−38 °C vs. −80 °C), which contributed to a faster and more reliable reaction. The reaction volume for the continuous process was estimated to be three orders of magnitude smaller than the batch process, which offers considerable safety benefits. Finally, scale-up of the process was achieved by ‘numbering-up’ the trickle-bed oxidation reactor. The researchers demonstrated that simply increasing the diameter (and hence the volume) of the reactor led to a dramatic loss of temperature control within the column and increased levels of impurities from the reaction were observed.
 | ||
| Scheme 3 Multi-kilogram scale synthesis of 6–hydroxybuspirone (LaPorte12). | ||
Synthetic chemists have long known that telescoping can be an effective tactic for truncating a multi-step synthesis.13 Telescoping reaction sequences typically involves the consecutive addition of reagents and/or catalysts to a reactor in order to initiate further transformations of intermediate products or to achieve in situ quenching of reactive species. This strategy is well suited to flow chemistry and a number of reports employing solution-based systems have been disclosed. Recently, the McQuade group reported a synthesis of the non-steroidal anti-inflammatory drug ibuprofen using continuous flow methods (Scheme 4).14 The three-step synthesis (Friedel–Crafts acylation, 1,2-migration and ester hydrolysis) was linked into a single continuous system and provided racemic ibuprofen in 51% isolated yield following off-line workup and crystallisation of the exiting flow stream. It is important to note that this multi-step telescoping sequence would be especially difficult (and unsafe!) to replicate using batch methods. The rapid change in reaction temperature (150 °C → 50 °C) would require considerable energy expenditure. Additionally, the pH adjustment from 1 → 14 is a highly exothermic reaction and is rendered manageable only by the high surface area and efficient heat transfer properties of the microreactor system.

The ability to perform multi-step reactions in an uninterrupted continuous fashion may also be beneficial for medicinal chemistry applications.15 Cosford recently described a continuous two-step synthesis of a focused 13-membered library of imidazo[1,2-a]pyridine-2-carboxamides (Scheme 5).16 No isolation of the carboxylic acid intermediates was required and a final off-line purification of the crude reaction mixture provided the targets. For their work the authors used the commercially available Syrris AFRICA flow system.17 For the example shown is Scheme 5 an impressive improvement in yield was observed for the flow synthesis compared to the original two-step batch method (46% vs. 16%).
 | ||
| Scheme 5 Synthesis of a Mur ligase inhibitor using multi-step continuous flow synthesis (Cosford16). | ||
Continuous separation and distillation
Although the telescoping processes described above are effective, they are not without limitations. A significant drawback is that excess reagents are often needed to quench leftovers from previous transformations, whilst the requirement for a holistic and careful route design (to ensure downstream reagent compatibility) is an added challenge. The integration of solution-based work-ups, with subsequent phase separation operations, into flow systems would therefore greatly expand the utility of this new technology.The Jensen group reported the integration of microfluidic biphasic extraction systems with microreactors for the multi-step synthesis of carbamates (Scheme 6).18 A microseparator incorporating a hydrophobic membrane was designed and used to successfully remove the aqueous stream and thus any water-soluble components.19

The Jensen group added a further instrument to the flow toolbox with the development of a microfluidic distillation unit capable of performing an in-line solvent switch. Working in conjunction with the Buchwald laboratory, a two-step flow sequence to prepare enol ethers was developed (Scheme 7).20 A bespoke silicon device was employed to carry out a continuous distillation of a binary solvent mixture (dichloromethane–DMF).21
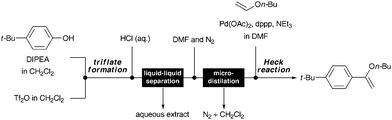 | ||
| Scheme 7 Continuous synthesis of an enol-ether involving liquid–liquid separation and continuous solvent exchange (Jensen and Buchwald20). | ||
Solid-supported multi-step flow synthesis
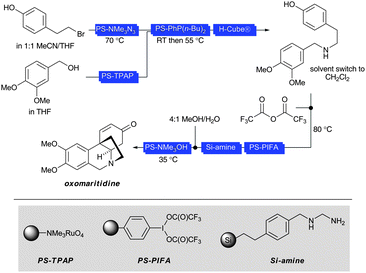
The use of supported reagents, catalysts and scavengers in synthesis is well documented and has proven to be an extremely advantageous technology in the modern laboratory.22 The combination of immobilized reagents with flow reactors23 has great potential for revolutionising the synthesis process.24The Ley group has pioneered the use of solid-supported reagents, catalysts and scavengers to facilitate organic synthesis and has an expanding portfolio of work in the area of continuous flow multi-step synthesis.25 Indeed, the group's 2006 synthesis of the complex natural product oxomaritidine is the most elaborate example of continuous flow multi-step synthesis to date (Scheme 8).26 Employing a variety of supported reagents, catalysts and scavengers, including the commercially available H-Cube hydrogenator,27seven synthetic steps were orchestrated into a single reactor network to afford the target in excellent yield (>40%). No traditional work-up or purification procedures were required to produce the natural product with >90% purity. Impressively, the entire sequence is completed in approximately six hours whilst performing the synthesis using traditional solid-phase assisted methods would take around four days of work. The labour savings are even more impressive when one considers that the system is automated, therefore releasing the researcher to undertake further tasks.
The development of catalytic processes is integral to the future of synthesis28 and so the use of solid-supported catalysts for multiple steps in flow systems is particularly attractive. Using an electroosmotic flow-driven miniaturized flow reactor, Watts recently reported the use of two solid-supported catalysts in series for the two-step synthesis of analytically pure α,β-unsaturated compounds (Scheme 9).29 The heterogeneous nature (and hence spatial-isolation) of the acidic and basic catalysts ensures their mutual compatibility and removes the need for any work-up or purification procedures.
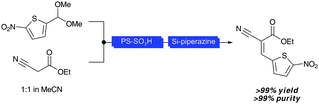 | ||
| Scheme 9 Continuous two-step synthesis of α,β-unsaturated compounds using supported catalysts (Watts29). | ||
In many instances, such as the synthesis of pharmaceuticals, the quality of the final product of a synthetic route must meet stringent purity standards. An effective method for achieving in-line purification in flow-mode is the integration of solid-supported scavengers to selectively remove unwanted components of the flow stream.
The Ley group recently reported on the multi-step synthesis of triazoles30 using the commercially available flow system from Vapourtec31 (Scheme 10). Following three chemical transformations (oxidation, homologation and ‘click’ triazole formation) the flowing solution was subsequently pumped through a variety of strategically positioned solid-supported scavengers. These scavengers effectively purged the flow stream of any leached copper (QP-TU), excess aldehyde (QP-BZA), basic (PS-SO3H) and acidic components (PS-NMe2), to provide the desired product in excellent purity and without recourse to traditional column chromatography.32
 | ||
| Scheme 10 Three-step continuous flow synthesis of a triazole employing a variety of immobilized reagents and scavengers (Ley30). | ||
The Lectka group has described the use of sequentially linked jacketed glass columns for catalytic and enantioselective multi-step flow synthesis and reported a continuous route to the metalloproteinase inhibitor BMS-275291 (Scheme 11).33 The use of scavenger columns eliminated the need for batch purification of the eluting flow stream. In their approach the flow streams were purely gravity-driven and Celite® was employed to control the reactor residence times. Remarkably impressive yields and selectivities were observed.
 | ||
| Scheme 11 Synthesis of BMS-275291 using a column-based system incorporating resin-bound reagents and scavengers (Lectka33). | ||
In a further example of a multiphase continuous flow system, Ulven reported the preparation of a 15-membered library of potential chemokine receptor ligands (Scheme 12).34 Three separate building blocks were combined in three distinct reaction steps, whilst two scavenger resins were employed to remove any unreacted substrates. Semi-automatic purification of the crude products allowed a high compound throughput, further underscoring the potential of continuous flow multi-step synthesis as a tool for the drug discovery process.
 | ||
| Scheme 12 Three-step continuous flow synthesis of receptor ligands (Ulven34). | ||
Finally, immobilized enzymes have also been integrated into continuous flow systems. Ley and co-workers reported the preparation of the natural product grossamide using a continuous flow reactor system (Scheme 13).35 An initial continuous peptide coupling protocol36 was followed by a peroxidase catalysed dimerization to deliver the natural product.
 | ||
| Scheme 13 A multi-step continuous synthesis of grossamide using an immobilised horseradish peroxidase (Ley35). | ||
Summary and outlook
In this mini-review we hope to have demonstrated that continuous flow multi-step organic synthesis is a burgeoning and exciting area of research. Furthermore, our own experiences in this area37 have suggested to us that this field has the potential to simplify and improve the synthesis process. Indeed, with the promise of economic, safety and time-saving benefits, pharmaceutical manufacturers have begun to investigate and implement continuous manufacturing as a viable alternative to the traditional batchwise synthesis of APIs.38 The ultimate goal may be to consolidate all aspects of manufacturing (synthesis, finishing and packaging) into a single location, thereby miniaturizing the entire production process.39However, despite much progress, in order to realise the full potential of flow chemistry a number of hurdles must still be overcome. The manipulation of solids, both as reagents and products, is still largely an unsolved problem and ‘clogging’ plagues much of the flow-based research currently being conducted.40 The assembly of several reactors in series often leads to a significant build-up of system pressure, requires the precise orchestration of multiple flow streams and can lead to prohibitively dilute reaction solutions. In these instances, continuous methods for solvent removal and switching become increasingly important. Recent reports21 of in-line distillation as part of a multi-step sequence are encouraging but may be limited to the removal of volatile solvents. A related technical challenge is reaction dispersion, particularly when heterogeneous systems are involved. As recently described by the Ley laboratory,21b, 41 when substrate dispersion becomes significant, controlling reagent stoichiometry for subsequent reaction steps becomes a considerable challenge. In-line and real-time analytical techniques may be a solution to this problem.41 Alternatively, catch-and-release strategies offer an ideal opportunity for product concentration, solvent switching and purification all in a single operation.42
Finally, a significant hurdle to the development of continuous flow organic synthesis may simply be unfamiliarity. Synthetic chemists are trained to conduct synthesis using traditional batch techniques and the technical aspects of flow chemistry may thus appear unnecessarily daunting and alien. Consequently, inter-disciplinary collaborations between chemists and engineers will be essential for future developments in continuous flow chemistry.43 Although many challenges remain, continuous flow multi-step synthesis may be a key breakthrough technology for enabling the efficient preparation of complex substances.
Acknowledgements
The authors would like to thank Novartis for its generous financial support of the Novartis-MIT Center for Continuous Manufacturing (CCM) and the members of the CCM for stimulating discussions, particularly Prof. Klavs F. Jensen, Prof. Stephen L. Buchwald, the members of their research groups and our Novartis colleagues Dr Berthold Schenkel, Dr Oljan Repic, Dr Thierry Schlama, Dr Mike Girgis and Dr Gerhard Penn. The authors would also like to thank Prof. S. V. Ley for helpful suggestions and comments.References
- For relevant discussions see: (a) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS; (b) P. T. Anastas and M. M. Kirchoff, Acc. Chem. Res., 2002, 35, 686 CrossRef CAS; (c) T. Hudlicky, Chem. Rev., 1996, 96, 3 CrossRef.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, Wiley, Chichester, 2009. For a recent review on biosynthetic approaches to total synthesis see Search PubMed; P. G. Bulger, S. K. Bagal and R. Marquez, Nat. Prod. Rep., 2008, 25, 254 Search PubMed.
- For a review on cascade reactions in total synthesis see: K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 Search PubMed; for C–H functionalisation methods see the thematic issue: Chem. Rev., 2010, 110, p. 575 CrossRef CAS; for a review on enantioselective cascade catalysis see: A. M. Walji and D. W. C. MacMillan, Synlett, 2007, 10, 1477 CrossRef CAS ; for protecting-group-free synthesis see: R.W. Hoffmann, Synthesis, 2006, 21, 3531 Search PubMed ; for a discussion of one-pot synthesis see: S. J. Broadwater, S. L. Roth, K. E. Price, M. Kobaslija and D. T. McQuade, Org. Biomol. Chem., 2005, 3, 2899 Search PubMed ; for multicomponent strategies see: B. B. Tour and D. G. Hall, Chem. Rev., 2009, 109, 4439 ; for an excellent demonstration of efficiency in synthesis see: K. B. Hansen, et al. , J. Am. Chem. Soc., 2009, 131, 8798 Search PubMed.
- For recent general reviews on continuous flow chemistry see: (a) T. Wirth, Microreactors in organic synthesis and catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) R. L. Hartman and K. F. Jensen, Lab Chip, 2009, 9, 2495 RSC; (c) B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300 CrossRef; (d) K. Geyer, T. Gustafsson and P. H. Seeberger, Synlett, 2009, 15, 2382; (e) A. Kirschning, W. Solodenko and K. Mennecke, Chem.–Eur. J., 2006, 12, 5972 CrossRef CAS; (f) C. Wiles and P. Watts, Eur. J. Org. Chem., 2008, 10, 1655 CrossRef; (g) S. V. Ley and I. R. Baxendale, Proceedings of Bosen Symposium, Systems Chemistry, 2008, 65 Search PubMed; (h) K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406 CrossRef.
- V. Hessel, Chem. Eng. Technol., 2009, 32, 1655 CrossRef CAS . Individual transformations that are conducted under continuous flow conditions (with non-continuous work-ups and purifications), albeit as part of a multi-step sequence, are beyond the scope of this mini-review. For example see: T. Schwalbe, D. Kadzimirsz and G. Jas, QSAR Comb. Sci., 2005, 24, 758 Search PubMed.
- In this mini-review we use the term ‘continuous flow synthesis’ to mean a synthetic process where chemical reactions are run using a continuously flowing stream. This definition is thus irrespective of the reactor type used and the scale involved. However, we acknowledge that reactor size is an important factor when discussing continuous flow synthesis. A thorough discussion of this topic is beyond the scope of this mini-review.
- J.-i. Yoshida, A. Nagaki and T. Yamada, Chem. Eur. J., 2008, 14, 7450 CrossRef CAS.
- H. Usutani, Y. Tomida, A. Nagaki, H. Okamoto, T. Nokami and J.-i. Yoshida, J. Am. Chem. Soc., 2007, 129, 3046 CrossRef CAS.
- In flow chemistry, residence time (tR) is the time that a reaction solution spends inside a reactor and is primarily a consequence of the flow rate.
- For a review on the large-scale use of organometallics in microreactors under flow conditions see: ref. 4(a) p211, and references cited within.
- T. Schwalbe, V. Autze, M. Hohmann and W. Stirner, Org. Process Res. Dev., 2004, 8, 440 Search PubMed.
- T. L. Laporte, M. Hamedi, J. S. DePue, L. Shen, D. Watson and D. Hsieh, Org. Process Res. Dev., 2008, 12, 956 Search PubMed.
- N. G. Anderson, Practical process research & development, Academic Press, San Diego, Calif.; London, 2000 Search PubMed.
- A. R. Bogdan, S. L. Poe, D. C. Kubis, S. J. Broadwater and D. T. McQuade, Angew. Chem., Int. Ed., 2009, 48, 8547 CrossRef CAS.
- For a discussion see: S. Y. F. Wong-Hawkes, J. C. Matteo, B. H. Warrington and J. D. White in New Avenues to Efficient Chemical Synthesis, 2007, Springer Berlin, Heidelberg, pp. 39–55 Search PubMed.
- A. Herath, R. Dahl and N. D. P. Cosford, Org. Lett., 2010, 12, 412 CrossRef CAS.
- Website: http://www.syrris.com/.
- H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704 CrossRef . For an alternative flow approach to carbamates, via azides, using solid-supported reagents see: M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin and C. D. Smith, Org. Biomol. Chem., 2008, 6, 1587 Search PubMed and M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin, C. D. Smith and J. P. Tierney, Org. Biomol. Chem., 2008, 6, 1577 RSC . For a very recent example of continuous multi-step synthesis incorporating continuous separations see: T. Tricotet and D. F. O'Shea, Chem. Eur. J., 2010, 16, 6678 Search PubMed.
- J. G. Kralj, H. R. Sahoo and K. F. Jensen, Lab Chip, 2007, 7, 256 RSC.
- R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 899 CAS.
- (a) R. L. Hartman, H. R. Sahoo, B. C. Yen and K. F. Jensen, Lab Chip, 2009, 9, 1843 RSC; (b) For a recently reported method for performing an in-line solvent switch as part of a multi-step flow sequence see: M. D. Hopkin, I. R. Baxendale and S. V. Ley, Chem. Commun., 2010, 46, 2450 Search PubMed.
- For a comprehensive compendium see: S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, J. Chem. Soc., Perkin Trans., 2000, 3815 Search PubMed . For other perspectives see: D. C. Sherrington, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2364 Search PubMed and P. Hodge, Chem. Soc. Rev., 1997, 26, 417 Search PubMed.
- For early discussions of multi-step flow synthesis using solid-supported reagents and catalysts see: (a) S. Ley, M. Bolli, B. Hinzen, A. Gervois, B. Hall, J. Habermann, J. Scott and F. Haunert, PCT Int. Appl., W09958475, 1999 Search PubMed; (b) S. V. Ley and I. R. Baxendale, Nat. Rev. Drug Discovery, 2002, 1, 573 CrossRef CAS; (c) G. Jas and A. Kirschning, Chem.–Eur. J., 2003, 9, 5708 CrossRef CAS; (d) P. Hodge, Curr. Opin. Chem. Biol., 2003, 7, 362 CrossRef CAS; (e) For further discussions see: I. R. Baxendale and S. V. Ley in New Avenues to Efficient Chemical Synthesis, 2007, Springer Berlin, Heidelberg, pp. 151–185 Search PubMed. For further discussions see references 4(e) and 22..
- P. Kundig, Science, 2006, 314, 430 CrossRef.
- For the most recent review of the group's work see reference 4(g).
- I. R. Baxendale, J. Deeley, C. M. Griffiths-Jones, S. V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 2566 RSC.
- Website: http://www.thalesnano.com/products/h-cube.
- R. Noyori, Nat. Chem., 2009, 1, 5 Search PubMed.
- C. Wiles, P. Watts and S. J. Haswell, Lab Chip, 2007, 7, 322 RSC . It is noteworthy that even ‘traditional’ synthesis laboratories have begun to embrace multi-step flow chemistry, see: A. W. Pilling, J. Boehmer and D. J. Dixon, Angew. Chem., Int. Ed., 2007, 46, 5428 Search PubMed.
- I. R. Baxendale, S. V. Ley, A. C. Mansfield and C. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 4017 CrossRef CAS.
- Website: http://www.vapourtec.co.uk/.
- For a discussion see: P. S. Seeberger, Nat. Chem., 2009, 1, 258 Search PubMed.
- S. France, D. Bernstein, A. Weatherwax and T. Lectka, Org. Lett., 2005, 7, 3009 CrossRef CAS . For a review of the Lectka group's work in this area see: A. M. Hafez, A. E. Taggi and T. Lectka, Chem. Eur. J., 2002, 8, 4114 Search PubMed.
- T. P. Petersen, A. Ritzen and T. Ulven, Org. Lett., 2009, 11, 5134 CrossRef CAS.
- I. R. Baxendale, C. M. Griffiths-Jones, S. V. Ley and G. K. Tranmer, Synlett, 2006, 3, 427.
- I. R. Baxendale, S. V. Ley, C. D. Smith and G. K. Tranmer, Chem. Commun., 2006, 4835 RSC.
- M. W. Bedore, N. Zaborenko, K. F. Jensen and T. F. Jamison, Org. Process Res. Dev., 2010, 14, 432 Search PubMed.
- D. M. Roberge, B. Zimmermann, F. Rainone, M. Gottsponer, M. Eyholzer and N. Kockmann, Org. Process Res. Dev., 2008, 12, 905 Search PubMed.
- C&E News, Oct. 8 2007, p.10 Search PubMed.
- For recent approaches to the handling of solids in continuous flow synthesis see: (a) T. Horie, M. Sumino, T. Tanaka, Y. Matsushiat, T. Ichimura and J.-i. Yoshida, Org. Process Res. Dev., 2010, 14, 405 Search PubMed; (b) S. L. Poe, M. A. Cummings, M. P. Haaf and D. T. McQuade, Angew. Chem., Int. Ed., 2006, 45, 1544 CrossRef CAS; (c) J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 12, 3618 Search PubMed.
- For the use of in-line IR analysis of continuous flow systems see: C. F. Carter, H. Lange, S. V. Ley, I. R. Baxendale, B. Wittkamp, J. G. Goode and N. L. Gaunt, Org. Process Res. Dev., 2010, 14, 393 Search PubMed , and references cited within.
- For a discussion of these concepts see ref. 23(b).
- http://engineering.mit.edu/research/labs_centers_programs/novartis.php .
| This journal is © The Royal Society of Chemistry 2010 |