A novel synergistic enhanced chemiluminescence achieved by a multiplex nanoprobe for biological applications combined with dual-amplification of magnetic nanoparticles†
Sai
Bi
,
Hong
Zhou
and
Shusheng
Zhang
*
Key Laboratory of Eco-chemical Engineering, Ministry of Education, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao, 266042, China. E-mail: shushzhang@126.com; Fax: +86 532 84022750; Tel: +86 532 84022750
First published on 7th October 2010
Abstract
A multiplex nanoprobe (CuS/DNA/Au/DNA/MNP) achieving a novel synergistic enhanced chemiluminescence (SECL) system is applied in a signal amplified bioassay strategy based on magnetic nanoparticles (MNPs) acting as both DNA molecular carriers and reporter labels, and further amplifying the signals by dual-amplification (DNA amplification and metal ions amplification). In this study, a luminol–H2O2–Cu2+–Fe3+ SECL system is proposed for the first time, which is achieved by a multicomponent nanoparticle probe and significantly intensifies the CL signal compared to monometallic Cu2+ or Fe3+ catalyst. The combination of the remarkable sensitivity of the SECL method with the amplification of MNPs achieves the detection of specific DNA sequences and Ramos cells as low as 6.8 aM and 56 cells mL−1, respectively, which is one of the most sensitive approaches for bioassays. In addition, the strategy is able to differentiate between Ramos target cells and CEM control cells based on the high specificity of the aptamer for Ramos cells, indicating the wide applicability of it for diseased cell detection.
Introduction
The emergence of nanotechnology is opening a new horizon for biological sensing, among which metal nanoparticles with distinctive photonic, magnetic and electronic properties offer an excellent prospect for highly sensitive assays of biological molecules.1–3 So far, two main approaches have been developed for nanoparticle-based bioassays. (1) Nanoparticles are used as carriers to load a large amount of species, e.g., enzyme molecular, DNA, and luminescent labels.4–6 As one of the most widely used nanomaterials in bioanalysis, magnetic nanoparticles (MNPs) are usually employed as special biomolecules immobilizing carriers because of their easy magnetic separation and large surface.7–10 (2) Metal nanoparticles are directly used as reporters. One strategy is based on the aggregation of oligonucleotide-functionalized gold nanoparticles by colorimetric detection for DNA hybridization, metal ions or proteins.11–13 Although this technique is easy to readout and rapid for detection, the low sensitivity limits its application. Another strategy of metal nanoparticle-based bioassay is to detect the metal ions released from the metal nanoparticle probes anchored on the reporters by oxidative metal dissolution.14,15As one of the most sensitive techniques for trace analysis of metal ions, chemiluminescence (CL) has become an attractive method for DNA and protein biosensing owing to its high sensitivity, inherent simplicity, and low cost.16–18 In previous reports, the CL system was only studied by catalyzing with individual metal ions, such as Au3+,17 Fe3+,18 or Cu2+.19 However, so far there has been no report on two metal ions acting simultaneously as catalysts for the CL reaction system. Inspired by the above two application fields of metal nanoparticles in biological analysis, we hypothesized that the MNPs-based protocol could produce a universal, selective and sensitive method for the collection and subsequent detection of various target molecules by combining the easy separation of MNPs as carriers, high immobilization as labels, and sensitive detection of metal ions released from MNPs by CL detection.
Herein, we make the best use of MNPs for biological applications based on a novel luminol–H2O2–Cu2+–Fe3+ SECL system by flow-injection chemiluminescence (FI-CL) detection, which is achieved from a CuS/DNA/Au/DNA/MNP multiplex nanoprobe and further accomplishes high sensitive and selective detection of specific DNA sequences. Meanwhile, MNPs are simultaneously acting as a dual-amplification intermediary for DNA amplification and metal ions amplification, allowing a detection limit of 6.8 aM for target DNA in DNA hybridization detection which is comparable with other most ultrasensitive DNA detection.20,21 Furthermore, based on the structure–switching of aptamers specific for Ramos cells selected from cell-SELEX upon target binding,22,23 the aptamer–ligand recognition event can be easily amplified following the nucleic acid sequence-based amplification protocols, which makes an extraordinarily low detection limit for Ramos cells possible. The remarkable sensitivity of the amplification strategy for the detection of Ramos cells can be as low as 56 cells mL−1, which is much more sensitive than other optical cytosensors.23,24 Given the excellent analytical properties and design flexibility, the developed strategy exhibits a promising analysis platform for diverse analytes assay. The preparations and analytical characteristics of the proposed DNA and whole-cell amplified assay schemes are detailed in this work.
Results and discussion
Principles of the proposed strategies for DNA and whole-cell amplified assay
In this study, MNPs were simultaneously acting as aptamer carriers and reporter labels, and further amplified the signals by employing MNPs as the dual-amplification intermediary based on the novel luminol–H2O2–Cu2+–Fe3+ SECL system achieved with a CuS/DNA/Au/DNA/MNP multiplex nanoprobe. Scheme 1A depicts the principle of dual-amplification strategy for target DNA (t-DNA) detection in DNA hybridization. Briefly, carboxyl modified MNPs (carboxyl-MNPs, ∼500 nm) were firstly functionalized with 5′-NH2 labeled bio-bar-code DNA (bbc-DNA) and first-probe DNA (p1-DNA), fabricating the bbc-p1-DNA/MNPs to avoid cross-linking reaction. Then, the streptavidin-coated substrate was modified with biotinylated capture DNA (biotin-c-DNA) (step I, Scheme 1A), followed by hybridizing with t-DNA on the substrate (step II, Scheme 1A). Subsequently, the bbc-p1-DNA/MNPs were conjugated onto the substrate by utilizing a hybridization reaction between p1-DNA and t-DNA (step III, Scheme 1A). In this case, only one MNP was bound to one t-DNA due to the effective avoidance of cross-reaction by employing bbc-p1-DNA/MNPs as labels, which was verified by hybridizing the CdTe quantum dots (CdTe QDs)-tagged probe DNA to bbc-p1-DNA/MNPs on the substrate. The centric distance between fluorescent spots indicated that one t-DNA could bring one MNP onto the substrate with a concentration of t-DNA of 1.0 × 10−14 M. Then, the bbc-p1-DNA/MNPs were released from the substrate via urea dehybridizaiton (step IV, Scheme 1A) and hybridized with the previously fabricated CuS NPs and Au NPs modified second-probe DNA (CuS/p2-DNA/Au) (step V, Scheme 1A). From Fig. 1B, the disappearance of fluorescence spots on the substrate indicated that the bbc-p1-DNA/MNPs were released from the substrate completely by a 50% (w/w) urea solution. It should be noted that in order to amplify the signals as large as possible, herein two kinds of second-probe DNA (p2-DNA), which were complementary to bbc-DNA and p1-DNA on the released MNPs surface, respectively, were employed. The nonspecifically bound nanoparticle probes could be automatically removed via magnetically controlled washing. Subsequently, once CuS NPs were dissolved from the hybrids by a certain concentration of nitric acid (0.1 M), a large amount of released cupric ions (Cu2+) could be sensitively determined by the luminol–H2O2 CL reaction system and generated a CL signal (step VI, Scheme 1A). The CL intensity was proportional to the concentration of t-DNA based on the amounts of dissolved Cu2+. Moreover, in order to amplify the CL signals at utmost, we dissolved ferric ions (Fe3+) of MNPs and Cu2+ of CuS NPs simultaneously via a higher concentration of nitric acid (0.4 M) (step VII, Scheme 1A) and surprisingly found the luminol–H2O2–Cu2+–Fe3+ SECL reaction system, whose sensitivity was approximately one order of magnitude higher than that just employing monometallic Cu2+ as catalyst, achieving a low detection limit of target DNA at 6.8 aM.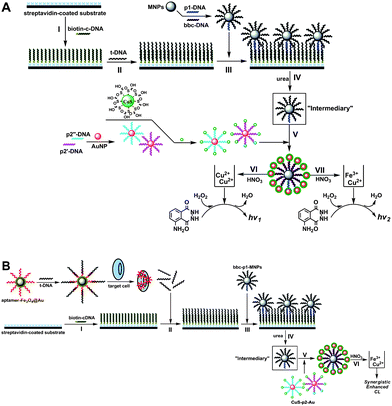 | ||
| Scheme 1 Schematic representation of SECL detection of DNA hybridization and Ramos target cells based on a dual-amplification strategy. | ||
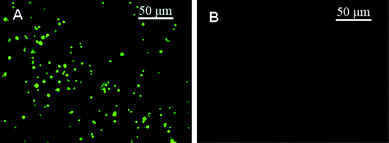 | ||
| Fig. 1 Images of the microtiter plate with CdTe QDs/bbc-p1-DNA/MNPs hybridized with 1.0 × 10−14 M t-DNA (A) before and (B) after dehybridization using 50% (w/w) urea solution. | ||
After investigating the mechanism of SECL system extensively (vide infra), a strategy for cancer cells detection was further developed based on the above proposed DNA-based amplification protocol. Scheme 1B displays the schematic diagram for Ramos cells detection. In this assay, amino-modified aptamers were attached on the surface of Fe3O4–Au core–shell magnetic nanoparticles (Fe3O4@Au) due to its large surface, and t-DNA, which was the same as that used in DNA hybridization detection with 12-mer complementary to the aptamer, was added to fabricate the t-aptamer-DNA/Fe3O4@Au complexes. The introduction of the Ramos target cells triggered the aptamers functionalized Fe3O4@Au assembling onto the cell surface through the conformational change of aptamers from a DNA/DNA duplex to a DNA/target complex. In this case, Fe3O4@Au-tagged target cells could be easily separated with a small magnet and again dispersed immediately after the magnet was removed. The released t-DNA was then detected according to the above amplified protocol for DNA hybridization detection by dissolving Cu2+ and Fe3+ simultaneously, whose signal was proportional to the concentration of Ramos cells.
Characterization of the DNA-NP conjugates
The synthesized Au NPs, CuS NPs, p2-DNA tagged with Au NPs and CuS NPs (CuS/p2-DNA/Au), and MNPs were characterized by TEM. From Fig. 2A–D, it can be seen clearly that in the absence of p2-DNA, only individual NPs were observed (A for Au NPs and B for CuS NPs, respectively), while in the presence of p2-DNA, aggregates of the Au NPs and CuS NPs were obtained (C). Fig. 2D shows the TEM image of the MNPs with an average diameter of 500 nm.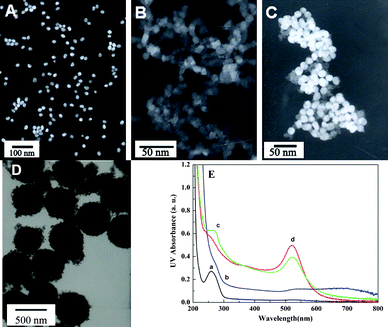 | ||
| Fig. 2 The TEM images of (A) AuNPs (∼20 nm), (B) CuS NPs (∼5 nm), (C) p2-DNA tagged with Au NPs and CuS NPs (CuS/p2-DNA/Au), and (D) MNPs (∼500 nm). (E) UV-visible spectra of (a) p2-DNA, (b) CuS NPs, (c) Au NPs, and (d) p2-DNA difunctionalized with CuS and Au NPs on the 3′- and 5′-ends. | ||
The UV-visible spectra of the CuS NPs, Au NPs, unmodified p2-DNA, and difunctionalized p2-DNA with CuS and Au NPs were recorded by the spectrophotometer as shown in Fig. 2E. Curve d exhibits both the characteristic absorbance of DNA (curve a) at 260 nm and Au NPs (curve c) at 520 nm. The results indicated that the CuS and Au NPs had been successfully labeled on the 3′- and 5′-end of p2-DNA, respectively.
DNA hybridization detection
In order to evaluate the feasibility of the demonstrated assay, the sensitivity of the DNA biosensor was first investigated. At step VIScheme 1A, once CuS NPs were dissolved from the hybrids, a large amount of released Cu2+ could be sensitively determined by the luminol–H2O2 CL reaction system, achieving a detection limit of 72 aM for t-DNA, which was comparable to that of 86 aM detected at step IVScheme 1A by the lunimol–H2O2–Fe3+ system. Here, t-DNA was 36-mer synthesized DNA sequence (sequence 3 in Table S1†).As an expansion study, we further dissolved CuS NPs and MNPs simultaneously after dehybridizing by urea solution and hybridizing with CuS/p2-DNA/Au via certain concentrations of nitric acid and detection by the luminol–H2O2–Cu2+–Fe3+ system with a desire to achieve a higher sensitivity. Surprisingly, in this study we found that Cu2+ and Fe3+, both luminol–H2O2 CL system catalysts, could significantly intensify the CL signals compared to monometallic Cu2+ or Fe3+ catalyst. The results showed that the CL intensity of the luminol–H2O2–Cu2+–Fe3+ CL system increased with increasing concentrations of t-DNA ranging from 1.0 × 10−17 to 1.0 × 10−15 M with a detection limit of 6.8 aM by using 3σ (σ = Sb/m; Sb, standard deviation of blank sample, Sb = 3.4801 (n = 11) in this experiment; m = 15.3532, the slope of the calibration curve at low concentration, 1.0 × 10−17 to 1.0 × 10−16 M in this experiment). A relative standard deviation (RSD) of 4.2% for eleven successive measurements of 4.0 × 10−17 M target DNA exhibited a good reproducibility of the assay.21 Consequently, the sensitivity of the present work was significantly improved based on the dual-amplification of MNPs and further improved by detecting dissolved Cu2+ and Fe3+ at the same time (Table S2†). The detection limit was comparable with other most ultrasensitive DNA detection.20,21 The mass concentration ratio of Cu2+ and Fe3+ dissolved from CuS/p2-DNA/Au/MNPs conjugates was 14/1. At this ratio, a synergistic effect between Cu2+ and Fe3+ for the luminol–H2O2 CL system was obtained, whose detection sensitivity was significantly improved compared to monometallic Cu2+ or Fe3+ as catalyst. In addition, the proposed amplification strategy was successfully applied in E. coli genomic DNA detection for 120- and 250-bp PCR products (sequences 11 and 12, respectively) by employing a new set of c-DNA (sequence 14), p1-DNA (sequence 15) and p2-DNA (sequence 16). The detailed results are shown in the Supporting Information.†
Investigation of luminol–H2O2–Cu2+–Fe3+ SECL
In order to reason the mechanism of the luminol–H2O2–Cu2+–Fe3+ SECL reaction system, the UV-visible absorption spectra and fluorescence spectra of different systems were investigated and nearly identical spectra were obtained (Fig. S8†). Thus, it could be proposed that no coordination complex was formed in the SECL system, and it was not the coordination complex mechanism. Subsequently, the effects of ·O2− quencher superoxide dimutase (SOD) and three HO· quenchers, mannitol, methanol and methyl benzoate, were investigated. As shown in Fig. 3A, the CL intensities were significantly quenched by these quenchers. Thus, it is reasonable that the possible mechanism of the luminol–H2O2–Cu2+–Fe3 SECL system was the “free radical mechanism” (Fig. 3B), in which a synergistic effect occurred between Cu2+ and Fe3+ in the generation of active oxygen radicals for the luminol–H2O2 CL system.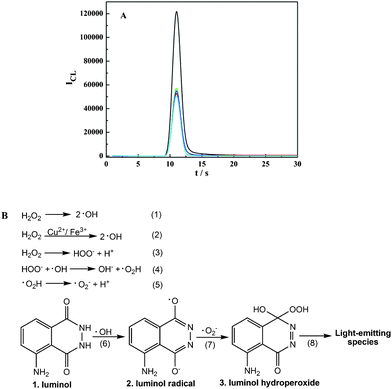 | ||
| Fig. 3 (A) Effect of quenchers on the mechanism of the luminol–H2O2–Cu2+–Fe3+ SECL system. 1.5 × 10−3 M luminol, 8.0 × 10−3 M H2O2, 1.0 × 10−8 g mL−1 Fe3+ and 1.4 × 10−7 g mL−1 Cu2+ were mixed and reacted with H2O (black), 100 μg mL−1 SOD (red), 0.1 M methanol (green), 0.01 M mannitol (blue), and 0.1 M methyl benzoate (cyan), respectively. (B) The presumptive mechanism of the luminol–H2O2–Cu2+–Fe3+ SECL system. | ||
In addition, for the general and wide application of the current SECL technique, the applicable ratios between Cu2+ and Fe3+ were also investigated. Considering the linear ranges of Cu2+ and Fe3+ in the luminol–H2O2 CL system were 1.0 × 10−9 to 1.0 × 10−8 g mL−1 and 1.0 × 10−10 to 1.0 × 10−9 g mL−1, respectively (Fig. S6 in Supporting Information†), in this study we set the concentration of Fe3+ as 1.0 × 10−11 g mL−1, at which the CL signal could not be detected in the luminol–H2O2–Fe3+ CL system, and investigated the ratios between Fe3+ and Cu2+ from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:500. Among the ratios from 1:1 to 1:100, the corresponding concentrations of Cu2+ were from 1.0 × 10−11 to 1.0 × 10−9 g mL−1 which could also not be detected in the luminol–H2O2–Cu2+system. Thus, if a CL signal could be measured in a Cu2+ and Fe3+ mixture for luminol–H2O2 CL system in this case, the novel SECL system could be proved. As shown in Fig. 4, when the ratios were from 1:1 and 1:50, both CL signals of the luminol–H2O2–Cu2+ (purple) and luminol–H2O2–Fe3+ (blue) CL systems could not be detected. Only if Fe3+ and Cu2+ simultaneously existed in the luminol–H2O2 CL system from a ratio of 1:5, could the CL signals be detected and SECL began to make sense. Furthermore, when the ratios between Fe3+ and Cu2+ were 1:100 and 1:500, the corresponding concentrations of Cu2+ were 1.0 × 10−9 and 5.0 × 10−9 g mL−1, the SECL still played a significant role in this case, although Cu2+ itself could be detected in the luminol–H2O2 CL system when the concentration of Cu2+ was higher than 1.0 × 10−9 g mL−1. Consequently, it is well-grounded that the SECL technique has been successfully proposed and is applicable when the ratios between Fe3+ and Cu2+ are from 1:5.
:1 to 1:500. Among the ratios from 1:1 to 1:100, the corresponding concentrations of Cu2+ were from 1.0 × 10−11 to 1.0 × 10−9 g mL−1 which could also not be detected in the luminol–H2O2–Cu2+system. Thus, if a CL signal could be measured in a Cu2+ and Fe3+ mixture for luminol–H2O2 CL system in this case, the novel SECL system could be proved. As shown in Fig. 4, when the ratios were from 1:1 and 1:50, both CL signals of the luminol–H2O2–Cu2+ (purple) and luminol–H2O2–Fe3+ (blue) CL systems could not be detected. Only if Fe3+ and Cu2+ simultaneously existed in the luminol–H2O2 CL system from a ratio of 1:5, could the CL signals be detected and SECL began to make sense. Furthermore, when the ratios between Fe3+ and Cu2+ were 1:100 and 1:500, the corresponding concentrations of Cu2+ were 1.0 × 10−9 and 5.0 × 10−9 g mL−1, the SECL still played a significant role in this case, although Cu2+ itself could be detected in the luminol–H2O2 CL system when the concentration of Cu2+ was higher than 1.0 × 10−9 g mL−1. Consequently, it is well-grounded that the SECL technique has been successfully proposed and is applicable when the ratios between Fe3+ and Cu2+ are from 1:5.
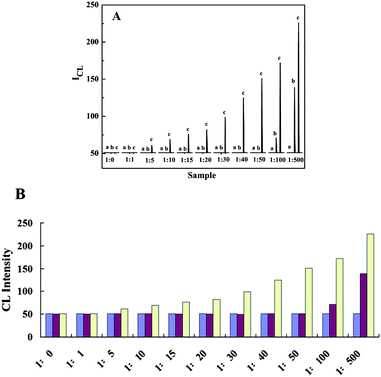 | ||
| Fig. 4 Investigation of the applicable ratios between Cu2+ and Fe3+ in the luminol–H2O2–Cu2+–Fe3+ SECL system. (A) Kinetic curves of FI-CL: (a) luminol–H2O2–Fe3+, (b) luminol–H2O2–Cu2+, and (c) luminol–H2O2–Cu2+–Fe3+. (B) Corresponding histogram: CL signals of luminol–H2O2–Fe3+ (blue), luminol–H2O2–Cu2+ (purple) and luminol–H2O2–Cu2+–Fe3+ (yellow). The concentration of Fe3+ is fixed as 1.0 × 10−11 g mL−1, and the concentrations of Cu2+ are changed corresponding to the ratios. The ratio of 1:0 represents that there is only Fe3+ and not Cu2+ in the luminol–H2O2 CL system. The baseline of the FI-CL is ∼50. | ||
Cancer cells detection
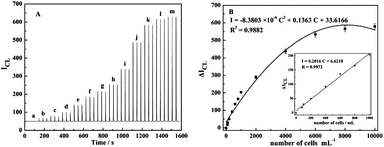 | ||
| Fig. 5 (A) FI-CL signals for Cu2+ and Fe3+ dissolved simultaneously from a series of CuS/p2-DNA/Au/MNPs conjugates corresponding to different concentrations of Ramos cells: (a) 0; (b) 80; (c) 100; (d) 200; (e) 400; (f) 600; (g) 800; (h) 1000; (i) 2000; (j) 4000; (k) 6000; (l) 8000; (m) 10000 cells mL−1. (B) The calibration curve of peak height versus the concentration of Ramos cells from 80 to 10000 cells mL−1. Inset is the amplification of the linear range from 80 to 1000 cells mL−1 for Ramos cells determination. | ||
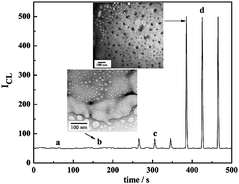 | ||
| Fig. 6 CL signals of (a) t-DNA/aptamer/Fe3O4@Au incubating without cells (blank), (b) t-DNA/aptamer/Fe3O4@Au incubating with CEM control cells, (c) t-DNA/random DNA/Fe3O4@Au incubating with Ramos target cells, and (d) t-DNA/aptamer/Fe3O4@Au incubating with Ramos target cells. The TEM images of aptamer/Fe3O4@Au assembled on target cell (sample d) and control cell (sample b). The concentration of each cell sample is 4000 cells mL−1. | ||
Mixed samples assay
In order to evaluate the potential application of the proposed method, determination of the detection capability of the assay was performed using artificial complex samples by mixing equal amounts of Ramos target cells and CEM control cells and imaged by confocal microscopy. In this experiment, CdTe QDs were used to label CEM cells prior to t-aptamer-Fe3O4@Au incubation in order to differentiate Ramos from CEM cells. CdTe-labeled CEM control cells were mixed with unlabeled Ramos cells with a ratio of 1:1 (Fig. 7A). After adding t-DNA/aptamer/Fe3O4@Au and incubating at 37 °C for 15 min with occasional gentle stirring, a magnetic field was applied to remove the control cells that were not attached to the aptamer-labeled Fe3O4@Au. The magnetically extracted and supernatant samples were then analyzed by confocal imaging as shown in Fig. 7B and C, respectively. The experiment was also performed by labeling Ramos target cells with CdTe QDs and mixing them (1:1) with unlabeled control cells as in Fig. 7D. The target cells separated by magnetic separation exhibited a CdTe fluorescence signal (Fig. 7E), while the supernatant control sample did not show fluorescence emission (Fig. 7F). The presence of the CdTe fluorescence proved that only the Ramos cells were collected and imaged. From the above results, it can be seen clearly that the assay was able to collect the Ramos cells in complex sample. The lack of the CdTe signal in Fig. 7B, along with the presence of the CdTe signal in Fig. 7E, proved that only target cells were binding collectedly using this method for extractions from 1:1 cell mixtures. The results were also confirmed by SECL strategy (Fig. S10†).
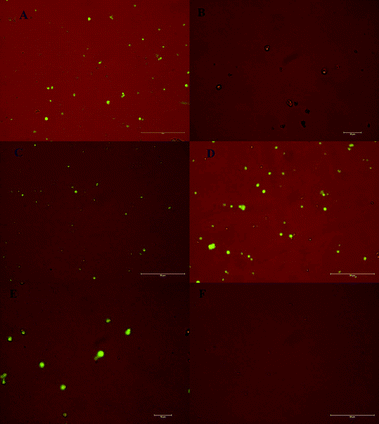 | ||
| Fig. 7 Images of (A) 1:1 ratio of target cells with CdTe QDs-stained control cells. After magnetic extraction, (B) target cells extracted by aptamer/Fe3O4@Au, and (C) CdTe QDs-stained control cells in supernatant. (D) 1:1 ratio of CdTe QDs-stained target cells with control cells. After magnetic extraction, (E) CdTe QDs-stained target cells extracted by aptamer/Fe3O4@Au, and (F) control cells in supernatant. Here, Ramos cells are taken as target cells, and CEM cells as control cells. | ||
Blood samples assay
Blood samples were also used to show the applicability of the method in a real assay of biological samples. In order to accomplish this end, Ramos cells were spiked into whole blood samples and compared with unspiked samples. After magnetic extraction, the stepwise double amplification SECL analysis process was performed as described above. Table S5† shows the SECL intensities of the blood samples with and without the defined added amounts of Ramos cells. The recovery was found to vary from 81.0 to 98.6%. Each sample was repeated three times and averaged to obtain the recovery values.Conclusions
In summary, the cooperative action between Cu2+ and Fe3+ led unique SECL system for luminol–H2O2 is achieved from a CuS/DNAAu/DNA/MNPs multiplex nanoparticle probe, which notably improves the sensitivity compared to that employing a monometallic ion as catalyst. Coupled with dual-amplified function of MNPs by DNA amplification and synergistically enhanced metal ions amplification, the method allows the detection of DNA down to 6.8 aM, which is comparable with other most ultrasensitive DNA detection.20,21 Moreover, for the detection of cancer cells, MNPs are also demonstrated to selectively extract Ramos target cells and significantly amplify FI-CL signals as reporter labels based on the cell-SELEX technique for aptamer selection. Whereas most previous studies for cells assay have to bind cells on supporters,28,29 this strategy is simpler and more flexible due to the selectivity and affinity of aptamers selected for Ramos cells. The variability in fabrication procedures, automation and succession of the FI-CL detection step, and good reproducibility of the method opens up a new avenue for sensitive and selective detection of a wide spectrum of aptamer-based analytes and holds great promise for direct analytical applications in vivo or in vitro.Experimental section
Synthesis of Au NPs
Au NPs were prepared according to the method of reduction of tetrachloroauric acid with trisodium citrate. Briefly, 100 mL of 0.01% HAuCl4 solution was heated to boiling with vigorous stirring, and then 3.0 mL of 1% trisodium citrate solution was added dropwise rapidly. After the color of the solution changed from gray yellow to deep red, the heating source was removed and the resulting gold colloidal suspension with an average diameter of ∼20 nm (0.6 nM) was stirred for an additional 15 min to cool down to room temperature.Preparation of water-soluble CuS NPs
In brief, 3.0 μL mercaptoacetic acid was added to 50 mL of 0.4 mM CuSO4 solution, and the pH of the mixture was adjusted to 9.0 with 0.5 M NaOH solution. After being bubbled with N2 for 30 min, 1.34 mM Na2S solution was added dropwise to the mixture to keep the molar ratio of Na2S to CuSO4 at approximately 2.5. The reaction was carried out for 24 h under N2, and a brown colloid was formed gradually.Modification of p2-DNA with Au NPs and CuS NPs
First, the p2-DNA/Au NPs were synthesized by dispersing 3 mL of Au NPs (0.6 nM) in 2 mL of 1.0 × 10−7 M 3′-thiol-modified p2′-DNA and 1.0 × 10−7 M 3′-thiol-modified p2′′-DNA, respectively, which were previously deprotected with TCEP for 1 h,25 followed by shaking gently for 16 h and “aged” in the solution (0.3 M NaCl, 10 mM Tris-acetate, pH 8.2) for another 48 h.26,27 Excess DNA was removed by successive washing of the Au NPs conjugates with centrifugation at 15 000 rpm for 30 min. The red oily precipitate was washed with 5 mL of 0.1 M pH 7.0 PBS buffer containing 0.1 M NaCl, recentrifuged, and dispersed in 5 mL of 0.1 M pH 7.0 PBS buffer solution containing 0.3 M NaCl. Approximately 25–30% of the original nanoparticle concentration was lost during each centrifugation and workup procedure.26 And then, 2.0 mL of 5′-amino group modified p2-DNA/Au solution was incubated with 200 μL of 0.1 M imidazole solution (pH 7.0) for 30 min, and dispersed in 100 μL of 0.1 M EDC solution and 2.0 mL of CuS colloid solution and stirred overnight. The resulting CuS/p2′-DNA/Au and CuS/p2′′-DNA/Au conjugates were centrifugated at 10 000 rpm for 30 min, followed by washing and resuspending in deionized and doubly distilled water. The two kinds of CuS/p2-DNA/Au (∼0.2 nM) were stored separately at 4 °C until use. The ratio of Au NPs, p2-DNA, and CuS NPs modified on the MNPs were ∼1:101:103.19Preparations of t-DNA/aptamer/Fe3O4@Au and bbc-p1-DNA/MNPs
The aptamer/Fe3O4@Au conjugates were synthesized by adding ∼50 μL of aqueous Fe3O4@Au solution (5 mg mL−1) to 500 μL of 2.0 × 10−8 M thiol-modified aptamer. Thiol-modified aptamer was deprotected and activated with TCEP (10 mM) for 1 h before attaching to Fe3O4@Au. After shaking gently for 16 h at room temperature, the aptamer-Fe3O4@Au conjugates were “aged” in the solution (0.3 M NaCl, 10 mM Tris-acetate, pH 8.2) for another 48 h. Excess reagents were removed by magnetic force, followed by adding 500 μL of 1.0 × 10−7 M t-DNA solution and incubating at 37 °C for 1 h to obtain the t-DNA/aptamer/Fe3O4@Au conjugates.The fabrication of bbc-p1-DNA/MNPs was carried out based on a slightly modified literature protocol.18 Briefly, 100 μL suspension of MNPs (10 mg mL−1) was transferred into a 1.5 mL Eppendorf (EP) tube. The MNPs were then washed three times with 400 μL of 0.1 M imidazol-HCl buffer (pH 7.0), and a 0.1 M imidazol-HCl buffer (300 μL) and a 0.8 M EDC (100 μL) were added to the EP tube. The mixture was incubated at 37 °C for 30 min to activate the carboxylate groups on the MNPs, followed by washing three times with 400 μL of 0.1 M PBS buffer. Then, 1.8 mL of 7.0 × 10−7 M bbc-DNA and 180 μL of 7.0 × 10−7 M p1-DNA were added and the mixture was incubated at 37 °C overnight with gentle mixing. Finally, the resulting MNPs, coated with bbc-DNA and p1-DNA, were washed three times with 400 μL of 0.1 M PBS buffer and resuspended in 200 μL PBS buffer for further use at 4 °C. Given the advantage of MNP in simple and complete magnetic separation, it could be considered that the MNP concentration was not lost during the modification procedures with the final concentration of ∼5 mg mL−1 in 200 μL PBS buffer. The surface coverage of bbc-DNA and p1-DNA on MNPs was determined according to UV-visible absorbance and fluorescence. The ratios of MNPs, t-DNA and bbc-DNA were approximately 1/1.69 × 105/1.22 × 106.18
DNA assay
Cell assay
Magnetic extraction was carried out by adding specified amounts of t-DNA/aptamer/Fe3O4@Au to each cell sample followed by incubating the mixture for 15 min at 37 °C. And then a magnetic field was introduced to the sample container. After the magnetic separation, 10 μL of the supernatant containing released t-DNA which was proportional to the amounts of target cells was decanted using a pipette followed by the above operations of the DNA assay.Acknowledgements
The work was supported by the Excellent Young Scientists Foundation of Shandong Province (JQ200805), the National Natural Science Foundation of China (20827005), and the National Basic Research Program of China (2010CB732404).Notes and references
- X.-M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 5, 912–920 Search PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS.
- R. Gill, M. Zayats and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 7602–7625 CrossRef CAS.
- A. Bajaj, S. Rana, O. R. Miranda, J. C. Yawe, D. J. Jerry, U. H. F. Bunz and V. M. Rotello, Chem. Sci., 2010, 1, 134–138 RSC.
- G.-F. Jie, P. Liu and S.-S. Zhang, Chem. Commun., 2010, 46, 1323–1325 RSC.
- G. D. Liu and Y. H. Lin, J. Am. Chem. Soc., 2007, 129, 10394–10401 CrossRef CAS.
- S. Bi, Y. M. Yan, X. Y. Yang and S. S. Zhang, Chem.–Eur. J., 2009, 15, 4704–4709 CrossRef CAS.
- S. Bi, S. Hao, L. Li and S. S. Zhang, Chem. Commun., 2010, 46, 6093–6095 RSC.
- J.-C. Liu, W.-J. Chen, C.-W. Li, K.-K. Tony Mong, P.-J. Tsai, T.-L. Tsai, Y. C. Lee and Y.-C. Chen, Analyst, 2009, 10, 2087–294 Search PubMed.
- Y.-M. Huh, E.-S. Lee, J.-H. Lee, Y.-W. Jun, P.-H. Kim, C.-O. Yun, J.-H. Kim, J.-S. Suh and J. Cheon, Adv. Mater., 2007, 19, 3109–3112 CrossRef CAS.
- R. Elghnian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS.
- C.-W. Liu, Y.-T. Hsieh, C.-C. Huang, Z.-H. Lin and H.-T. Chang, Chem. Commun., 2008, 2242–2244 RSC.
- J. W. Liu and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8634–8643 CrossRef CAS.
- J. Wang, D. Xu, A.-N. Kawde and R. Polsky, Anal. Chem., 2001, 73, 5576–5581 CrossRef CAS.
- J. Wang, R. Polsky, A. Merkoci and K. L. Turner, Langmuir, 2003, 19, 989–991 CrossRef CAS.
- K. Aslan and C. D. Geddes, Chem. Soc. Rev., 2009, 38, 2556–2564 RSC.
- A. P. Fan, C. Lau and J. Z. Lu, Anal. Chem., 2005, 77, 3238–3242 CrossRef CAS.
- S. Bi, H. Zhou and S. Zhang, Chem. Commun., 2009, 5567–5569 RSC.
- S. Zhang, H. Zhong and C. Ding, Anal. Chem., 2008, 80, 7206–7212 CrossRef CAS.
- C. S. Thaxton, H. D. Hill, D. G. Georganopoulou, S. I. Stoeva and C. A. Mirkin, Anal. Chem., 2005, 77, 8174–8178 CrossRef CAS.
- B. Munge, G. Liu, G. Collins and J. Wang, Anal. Chem., 2005, 77, 4662–4666 CrossRef CAS.
- Z. Tang, D. Shangguan, K. Wang, H. Shi, K. Sefah, P. Mallikratchy, H. W. Chen, Y. Li and W. Tan, Anal. Chem., 2007, 79, 4900–4907 CrossRef CAS.
- C. D. Medley, J. E. Smith, Z. Tang, Y. Wu, S. Bamrungsap and W. Tan, Anal. Chem., 2008, 80, 1067–1072 CrossRef CAS.
- J. E. Smith, C. D. Medley, Z. Tang, D. Shangguan, C. Lofton and W. Tan, Anal. Chem., 2007, 79, 3075–3082 CrossRef CAS.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2005, 127, 12677–12683 CrossRef CAS.
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- J. Zhang, S. Song, L. Zhang, L. Wang, H. Wu, D. Pan and C. Fan, J. Am. Chem. Soc., 2006, 128, 8575–8580 CrossRef CAS.
- C. Hao, L. Ding, X. Zhang and H. Ju, Anal. Chem., 2007, 79, 4442–4447 CrossRef CAS.
- E. Han, L. Ding, H. Lian and H. Ju, Chem. Commun., 2010, 46, 5446–5448 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental section, DNA hybridization assays, investigation of SECL system, cell assays of mixed samples and blood samples, respectively. See DOI: 10.1039/c0sc00341g |
| This journal is © The Royal Society of Chemistry 2010 |