Charge stabilisation by dimer formation of an endcapped thiophene tetramer—an in situ NMR spectroelectrochemical study
Sabrina
Klod
a,
Kinga
Haubner
ab,
Evelin
Jähne
b and
Lothar
Dunsch
*a
aCenter of Spectroelectrochemistry, Department of Electrochemistry and Conducting Polymers, IFW Dresden, Institute of Solid State Research, Helmholtzstrasse 20, 01069, Dresden, Germany. E-mail: L.Dunsch@ifw-dresden.de; Fax: (+)49 351 4659 811
bChemistry Department, Macromolecular Chemistry, TU Dresden, Zellescher Weg 19, 01069, Dresden, Germany
First published on 4th October 2010
Abstract
The role of dimers in the charge stabilisation of 5,5′′′-dicyano-3,3′′-dibutyl 2,2′:5′,2′′:5′′,2′′′-quaterthiophene (DCNDBQT) via an electrochemical reaction is studied in detail by in situ NMR spectroelectrochemistry. Among the two oxidation steps the first was found to be reversible while the second is completely irreversible. By in situ NMR spectroelectrochemistry we are able to demonstrate the formation of a π-dimer via the cation generated in the first electron transfer as well as the formation of a σ-dimer by a follow-up reaction of the dication generated in the second oxidation step. As the thiophene oligomer is blocked in the 2- and 5-positions by the cyanide group the monomer undergoes a dimerisation in the 4′-position. In situ NMR spectroelectrochemistry delivers proof of the coupling reaction of the thiophene rings in the electrochemistry of α,ω-endcapped oligothiophenes although such endcapped structures are expected to be prevented from chemical follow-up reactions. Furthermore, substitution by cyano groups opens the route to a cathodic reduction of the structure. The dicyanodibutylquaterthiophene gives a single reduction step which turns out to be quasi-reversible but the anionic structure formed in the reduction was not detectable by in situ NMR spectroelectrochemistry. This and the follow-up reactions are discussed in detail.
Introduction
Functional oligothiophenes, with their large variety of mechanical, electronic, optical and redox properties due to their structural variation,1 are good candidates for applications in organic field-effect transistors (OFET)2 organic light emitting diodes (OLED),3 and organic solar cells.4 While a majority of studies deals with the preparation and characterisation of hole transport (p-type) materials only a few papers are focused on electron-transporting (n-type) structures which can be used as ambipolar organic devices. Until now changes in the structure of such compounds during electrochemical charge injection have not been discussed in detail.In situ spectroelectrochemistry comprises powerful methods for structure determination of intermediates and final products in electrochemical reactions.5 Especially the combination of in situ ESR spectroelectrochemistry with UV/Vis/NIR spectroscopy has been shown to open new routes for the determination of charged states in polymers and oligomers.6 For α-endcapped oligothiophenes stable monocation radicals are formed at longer homologues. The reversible formation of a π-dimer from the monocations is proposed based on temperature dependent ESR measurements pointing to the stabilisation of the formed radical via dimerisation. Short thiophene chains give rather reactive monocation radicals. Here the formation of a σ-dimer in the 5-position is proposed.8 Longer thiophene chains show the formation of a dimer in the second oxidation step.9 5-substituted oligothiophenes and α,ω-endcapped oligomers form stable mono- and dications, although a dimerisation in the 3-position is observed to a small extent, which was shown to be reversible.10
Addition of electron-withdrawing groups (CHO, CN, NO2, etc.) or their combination with donating substituents (alkyl, alkoxy, alkylamino, etc.) results in a tuning of the electronic properties of oligomers and provides a broader field for applications.2,11–13 The synthesis and electrochemical behaviour of cyano-substituted oligothiophenes published in the 1990s was limited to small (bi- and ter-) thiophenes.14–16 Later on spectro- and electrochemical studies of higher oligothiophenes end-capped by the nitrile group were reported using cyclic voltammetry, chronoamperometry, UV/Vis/NIR, IR, Raman and ESR spectroscopies.17–19
Although NMR spectroscopy plays an important role in the determination of rather small structural changes in molecules20 its combination with electrochemistry has rarely been used due to more experimental difficulties when compared to other spectroscopic methods.21 Because of the changes in probe head geometry by the introduction of conducting electrodes and of the strong magnetic field, such measurements are nearly impossible. Special requirements have to be considered for successful NMR spectroelectrochemistry. Symmetrically arranged carbon fibres combined with a silver wire as a pseudo reference provide an electrode system of large scale, thus making in situ NMR spectroelectrochemistry sensitive.22
In the present work we investigated the redox behaviour of DCNDBQT by cyclic voltammetry and in situ NMR spectroelectrochemistry. Our approach was to design an oligomer of good solubility in organic solvents by introduction of a short alkyl chain in the 3,3′′′-position and of high stability in the charged state by endcapping. In order to gain a more detailed insight into the electrochemical reaction mechanism in situ NMR spectroelectrochemistry was used.
Results and discussion
The electrochemical properties of DCNDBQT were studied by both cyclic (CV) and square-wave (SWV) voltammetries (Fig. 1, 2). All redox peak potentials (vs. DmFc/DmFc+) of the oligothiophene are given in Table 1.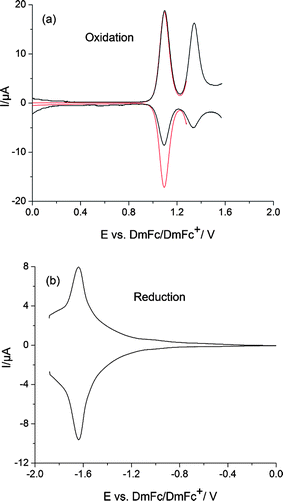 | ||
| Fig. 1 Square-wave voltammograms of DCNDBQT in ACN/TBAPF6; (a) oxidation, red: first oxidation step, black: first and second oxidation steps, (b) reduction. (Scan rate: 0.05 V s−1, concentration: 0.0005 mol l−1, working electrode: platinum wire.) | ||
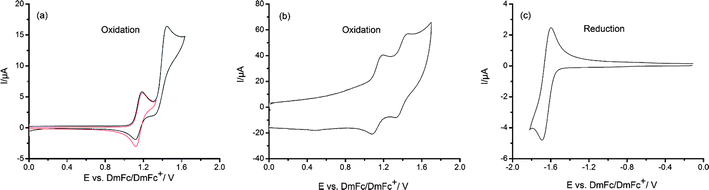 | ||
| Fig. 2 Cyclic voltammograms of DCNDBQT in ACN/TBAPF6: (a) oxidation, red: first oxidation step, black: first and second oxidation steps, scan rate: 0.05 V s−1, (b) oxidation, scan rate: 100 V s−1 (c) reduction, scan rate: 0.05 V s−1. (Concentration: 0.0005 mol l−1, working electrode: platinum wire.) | ||
| E pa/V | E pc/V | ΔEp/mV | E ½/V | |
|---|---|---|---|---|
| a E pc, Epa: cathodic and anodic peak potential, (scan rate 0.05 V s−1), ΔEp: peak potential separations, E½: half wave potential; * reversible in CV at 100 V s−1. | ||||
| Oxidation step (1st) | 1.19 | 1.12 | 70 | 1.15 |
| Oxidation step (2nd) | 1.44 | (1.33*) | — | — |
| Reduction step | −1.59 | −1.68 | 90 | −1.63 |
DCNDBQT displays two oxidation steps: the first at 1.15 V shows a peak potential separation ΔE1 = 70 mV pointing to a reversible electrochemical behaviour, while the second at 1.44 V turns out to be irreversible. Thus the first oxidation process with the reversible one-electron transfer results in an oxidation of the neutral molecule to the radical cation (DCNDBQT+). The irreversible second oxidation step is caused by chemical reactions following the electron transfer. The reversibility of the second oxidation, corresponding to the formation of the dication, is visible in cyclic voltammetry at higher scan rates (100 V s−1) when the very reactive charged structures will be re-reduced. From the anodic behaviour of DCNDBQT it can be concluded that the single positive charge is stabilized at the oligothiophene while doubly oxidized states are not stable at room temperature.
As cyano groups have electron acceptor properties the reduction of oligomeric structures of thiophenes are promoted. Therefore, contrary to unsubstituted oligothiophenes, DCNDBQT exhibits one quasi-reversible reduction step at −1.63 V. Following the reactions of both the higher anodic and cathodic processes shown to be irreversible in cyclic voltammetry requires spectroelectrochemical studies.
For spectroelectrochemical studies the NMR pattern of the redox systems in its virgin state has to be characterised first. The 1H NMR spectrum of DCNDBQT (Fig. 3a) consists of 7 independent signals. The signals at 0.95 ppm, 1.41 ppm, 1.65 ppm and 2.81 ppm are attributed to the aliphatic side group with intensities and couplings typical for the n-butyl group. In the range of aromatic protons two doublets at 7.19 ppm and 7.24 ppm and a singlet at 7.49 ppm can be found. They are attributed by their intensities and couplings to one isolated thiophene proton in the 4-position and two coupled thiophene protons in positions 4′ and 3′ (Scheme 1).
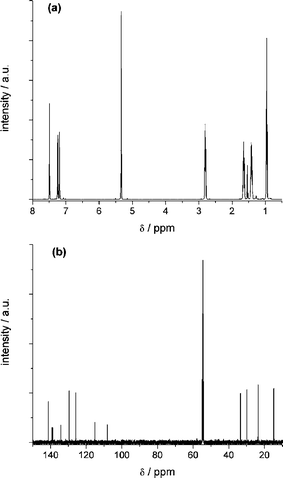 | ||
| Fig. 3 1H (a) and 13C (b) NMR spectra of DCNDBQT in CD2Cl2 obtained at 500 MHz and 125 MHz, respectively. | ||
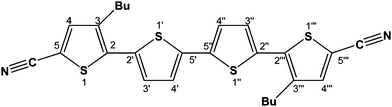 | ||
| Scheme 1 Chemical structure of 5,5′′′-dicyano-3,3′′′-dibutyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (DCNDBQT) | ||
The 13C NMR spectrum (Fig. 3b) shows the four signals expected of the aliphatic carbons in the n-butyl group at 14.66 ppm, 23.51 ppm, 29.84 ppm and 33.44 ppm, respectively. In addition, three NMR signals of higher intensity at 128.84 ppm, 129.62 ppm and 141.21 ppm and six signals of very low intensity at 108.13 ppm, 115.16 ppm, 134.15 ppm, 138.68 ppm, 139.16 ppm and 141.34 ppm are found. The signals of higher intensity can be attributed to the proton-bearing carbons in the thiophene ring chain in positions 4, 3′ and 4′ (Scheme 1), whereas the less intensive signals are attributed to carbons with substituents. The number of signals and their intensities confirm the proposed symmetry axis in the molecule.
NMR spectroelectrochemistry was applied to both oxidation steps following the structural changes under potentiostatic electrolysis. Taking into account the very long measuring time of NMR spectroscopy compared to cyclic voltammetry reactive and unstable species formed by electrochemical reactions are spectroscopically not detectable and only stable structures can be measured. Thus, the conclusions from in situ NMR spectroelectrochemistry cannot be analysed in this respect. Another point is the sensitivity of the NMR experiment. While in electrochemistry very low concentrations of species can be detected by the charge transferred in their electrochemical response, this is not the case in NMR spectroscopy. As NMR measurements have a cutoff concentration for the detection of species in solution, much lower concentrations of intermediates or byproducts are not detectable. Furthermore, in in situ NMR experiments the lower signal-to-noise ratio due to the filling of the tube with the electrode system leads to a lower sensitivity.22
Starting with the first oxidation peak at 1190 mV the NMR signal was analysed in detail. As DCNDBQT is only slightly soluble in acetonitrile its concentration is low resulting in a very low signal-to-noise ratio in the NMR spectrum.
While at an open cell potential condition (OCP) the NMR signals of the aromatic part in the thiophene oligomer are clearly detectable (Fig. 4a, the signals of the butyl group overlap with those of the supporting electrolyte and are therefore ignored) during electrolysis at 1190 mV its proton signals decrease to zero after 3 h. The dependence of the signal intensity on the charge is shown in Fig. 4 (upper part). This decrease is caused by the formation of a paramagnetic structure which is not detectable in NMR. Additional signals arise in that potential range. Because of the low concentration the intensity of the new signal is very small and cannot be clearly evaluated under these conditions. Reversing the potential scan to the open cell potential (see in Fig. 4b) the signals of DCNDBQT re-appear but the additional small signals arising upon oxidation do not disappear. Therefore the paramagnetic product is transformed into the neutral state but the additional structure as a consequence of a follow up reaction is not re-reduced.
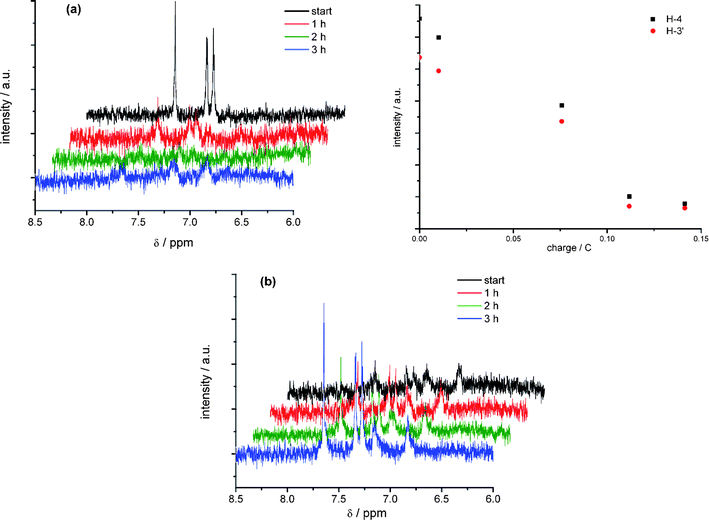 | ||
| Fig. 4 In situ 1H NMR spectroelectrochemistry of the oxidation (a) and correlation of charge and signal intensity during electrolysis, (b) back scan of DCNDBQT in acetonitrile/TBAPF6 at an electrode potential of 1190 mV (working electrode: carbon fibre). | ||
Temperature dependent ESR measurements of different types of oligothiophenes have shown the reversible formation of a π-dimer of radical cations in acetonitrile.6,7 This formation is confirmed from the vanishing of the ESR signal due to the formation of a diamagnetic dimer. Therefore we used temperature-dependent in situ NMR spectroelectrochemical measurements to follow the formation of a π-dimer of the DCNDBQT cation radical. Fig. 5 shows the in situ NMR spectroelectrochemical measurements at three different temperatures. While at 293 K no NMR signal is observed, a NMR signal similar to neutral DCNDBQT is found at 283 K and 273 K and is shifted by 0.1 ppm to lower field. This signal can be attributed to the diamagnetic π-dimer of the monocation radical of DCNDBQT. The slight shift of the signal to lower field is due to the lower electron density of the oxidised molecule.
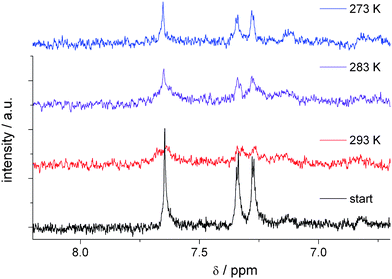 | ||
| Fig. 5 In situ 1H-NMR spectroelectrochemistry of DCNDBQT in acetonitrile/TBAPF6 at an electrode potential of 1190 mV (working electrode: carbon fibre) at three different temperatures. | ||
At the second anodic electron transfer the NMR signals of DCNDBQT vanish completely after extended electrolysis in acetonitrile solution (Fig. 6a). The NMR spectrum of the dicationic thiophene oligomer is not detectable. Reversing the potential to the open cell potential condition (Fig. 6b) no NMR signal of the neutral oligomer arises, pointing to an irreversibility caused by a chemical reaction which was already demonstrated by CV. As a precipitate of the follow-up product of the dicationic structure was found, the solution was changed to CD2Cl2 with TBAPF6 as the supporting electrolyte. Due to the lower conductivity of this solution and the lower current a longer electrolysis time is required.
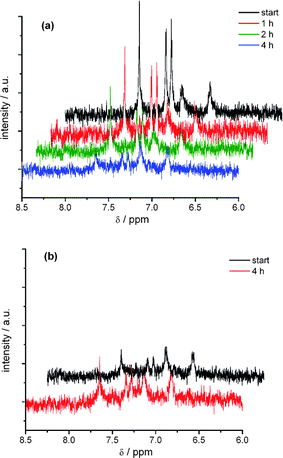 | ||
| Fig. 6 In situ 1H NMR spectroelectrochemistry of the oxidation (a) and back scan (b) of DCNDBQT in acetonitrile with TBAPF6 at an electrode potential of 1440 mV (working electrode: carbon fibre). | ||
With the better solubility of DCNDBQT in CD2Cl2 and a higher signal-to-noise ratio (cf.Fig. 7a) the analysis of the electrolysis product in CD2Cl2 after 12 h is possible. While, as in acetonitrile, the same decrease of signal intensity to about half confirms the conclusion of the experiments, additional signals arise in the NMR spectrum at 7.12 ppm (singlet) and 7.44 ppm (doublet). After reversing the potential to the OCP the signals of DCNDBQT increase (see Fig. 7b). Obviously the paramagnetic cation radical is formed in the methylenechloride solution as well which is re-oxidised in CD2Cl2 to the initial diamagnetic molecule. The formation of the additional product is also found under these conditions.
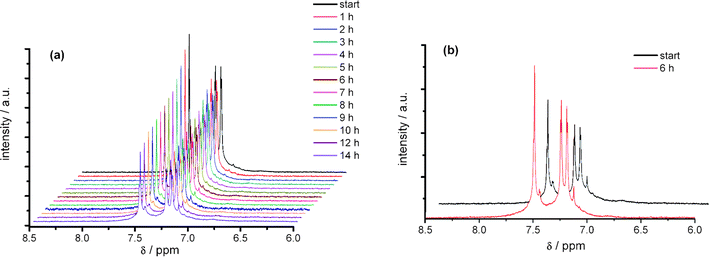 | ||
| Fig. 7 In situ 1H NMR spectroelectrochemistry of the oxidation (a) and the back scan (b) of DCNDBQT in CD2Cl2 with TBAPF6 at an electrode potential of 1190 mV (working electrode: carbon fibre). | ||
The electrolysis at the potential of the second anodic electron transfer (Fig. 8) leads to a decrease in intensity of the signals of DCNDBQT. The two additional signals, also obtained at the first electron transfer, arise and increase in intensity during this electrolysis. After vanishing of the DCNDBQT signal a pattern of three doublets and four singlets is detectable (Fig. 9).
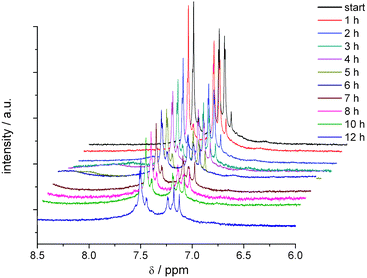 | ||
| Fig. 8 In situ 1H NMR spectroelectrochemistry of the oxidation of DCNDBQT in CD2Cl2 with TBAPF6 at an electrode potential of 1440 mV (working electrode: carbon fibre). | ||
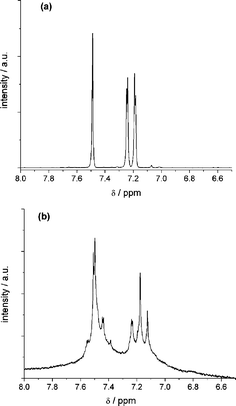 | ||
| Fig. 9 1H NMR spectra of the monomer (a) and the dimer (b) (only aromatic range is shown). | ||
This pattern can be attributed to a dimer formed by σ-bond formation between two thiophene rings in the 4′ position (cf.Scheme 1). The doubly charged DCNDBQT dimerises as the four thiophene rings cannot stabilise two charges. Therefore the dicationic structure is not stable. As the charged structure is stabilised by a dimer the extended conjugated chain is sufficiently large to stabilise the charge. The mechanism of dimer formation by thiophene ring coupling9,10 is shown in Scheme 2.
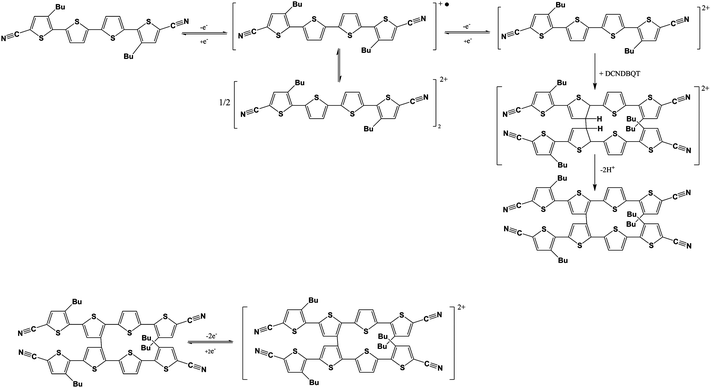 | ||
| Scheme 2 Mechanism of the oxidation of DCNDBQT including the chemical follow-up reactions via π- and σ-dimer formation. | ||
The coupling of the thiophene rings is accompanied by a proton abstraction. The dimerisation is not reversible in re-reduction. As mentioned in the literature10 oligothiophenes blocked in the 2- and 5- positions tend to dimerise in the 4-position. In DCNDBQT the dimer is formed via the 4′-position due to the shielding of the 4- and 3′-positions by the sterically extended butyl group. The dimer formed has a lower solubility as compared to the monomer, causing a precipitation. A brownish coloured precipitate is observable in the spectroelectrochemical cell after electrolysis. The NMR signals attributed to the σ-dimer were also detectable to some extent in the electrolysis at the first oxidation potential due to a potential gradient at the working electrode in the spectroelectrochemical cell.
Upon reduction a redox peak of DCNDBQT is found at a potential of −1680 mV. At this potential the formation of an anion radical is expected but the quasi-reversibility of the reduction step points to follow up reactions or at least the formation of some side products. The in situ NMR spectroelectrochemical experiment (Fig. 10a) shows a decrease of the signal intensity during electrolysis at −1680 mV. After 12 h the signal intensity is reduced to 5 percent. No additional signals arise in the NMR spectrum. Thus the formation of a paramagnetic structure is expected for a one-electron transfer. A second electron transfer would lead to the dianion being detectable as a diamagnetic structure by NMR spectroscopy.
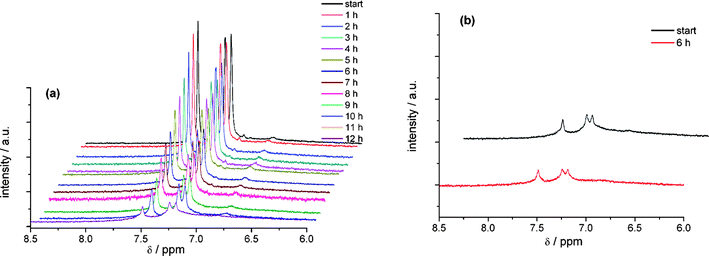 | ||
| Fig. 10 In situ 1H NMR spectroelectrochemistry of the cathodic reduction (a) and the back scan (b) of DCNDBQT in CD2Cl2 with TBAPF6 at an electrode potential of −1680 mV (working electrode: carbon fibre). | ||
If the electrode potential is reversed to the initial potential value the NMR signal does not re-appear (see Fig. 10b). Therefore a follow-up reaction of the formed paramagnetic structure to a product with very low solubility is to be assumed. After electrolysis a dark coloured precipitate is observed in the spectroelectrochemical cell. The colour of the formed product is different from the dimer formed by oxidation.
The precipitates of the electrochemical reactions were analysed by MALDI-TOF mass spectrometry. Both the precipitates from the anodic and the cathodic reactions as well as the DCNDBQT starting material were studied in 2,5-dihydroxybenzoic acid as a matrix and measured in the negative reflection mode. DCNDBQT shows a mass peak of 492 and a characteristic pattern of fragmentation. The mass spectrum of the oxidation product differs from the starting material by an additional peak at 984 with a similar isotope pattern which confirms the σ-dimer formation by follow-up chemical reaction of the highly oxidized quarterthiophene.
The reduction product shows, besides these two masses of the monomer and the dimer, a higher mass peak at 1476 pointing to the formation of a trimer or doubly charged hexamer due to a chemical follow-up reaction initiated by electrochemical reduction. Therefore the reduction of DCNDBQT results in polymeric structure formed by a follow-up reaction of the anion radical.
Experimental
Commercially available acetonitrile (ACN, puriss.; absolute; w(H2O) ≤ 0.001%) (Aldrich), toluene (Aldrich), dichloromethane (Aldrich), tetrahydrofuran (THF) from Acros, decamethylferrocene (p.a., ≥98.0%) purchased from Merck and 2,5-dihydroxy benzoic acid (2,5-DHB) from Aldrich were used without further purification. Tetrabutylammonium hexafluorophosphate (TBAPF6) of puriss. quality (Fluka) was dried under reduced pressure at 70 °C for 24 h and stored in a glove box.Cyclic (CV) and square-wave voltammograms (SWV) were measured in acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as supporting electrolyte using a one-compartment electrochemical cell with a platinum wire as working and counter electrodes and a Ag wire as a pseudo-reference electrode. DCNDBQT was dissolved at a concentration of 0.0005 mol l−1 in acetonitrile or 0.05 mol l−1 in CH2Cl2 and the deuterated solvents for the NMR experiments. Electrochemical measurements were performed at 22 ± 1 °C under nitrogen atmosphere. Data were recorded on an EG & G PAR 273A computer-controlled potentiostat. Decamethylferrocene was used as an internal reference. Cyclic voltammetry with scan rates higher than 50 V s−1 were recorded using an Autolab electrochemical analyzer equipped with a PGSTAT 100 potentiostat module and a fast analog scan generator (SCAN 250) in combination with a fast AD convertor (ADC10M). The working electrode area was minimized to 0.25 mm2.
MALDI-TOF measurements were carried out using a Bruker BIFLEX II equipped with a 337 nm nitrogen laser. The MALDI-TOF-spectra were measured in reflective negative mode. Samples were prepared by the sandwich method. First a layer of matrix solution 2,5-DHB in THF was added. After evaporation of the solvent, a second layer of oligothiophene solution in toluene was added and then the last, third, layer as a further matrix was loaded on the target. The solvent was removed by evaporation in air, and the sample transferred to the mass spectrometer for analysis. Mass spectra were calibrated by the fullerene C60 in a sulfur matrix.
The NMR measurements were performed on a Bruker Avance II 500MHz equipped with a standard 5mm BBO probe head, a heating system BC-U05 and the software package TopSpin 2.1 (Bruker Biospin Germany). A standard pulse sequence was used to obtain 1H and 13C spectra. The spectra were calibrated using the signal of the lock solvents acetonitrile-D3 or CD2Cl2, respectively.
For in situ NMR electrochemical experiments an electrode system consisting of carbon fibres as the working and the counter electrode were attached via copper wires to a PG 390USB potentiostat combined with an ITC 16 interface of Instrutech and using the software package Potpulse/Potmaster (all of HEKA Germany). The potential was either varied in steps or kept constant at a selected value. The experimental details of this NMR spectroelectrochemical method as well as the special cell design for this in situ method have been described elsewhere.22
3,3′′′-dibutyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (DBQT) was synthesized from 3-butyl-substituted-2-bromthiophenes by Grignard reaction followed by a coupling with commercially available dithienyldibromide. Resulting DBQT was brominated by NBS to form the corresponding 5,5′′′-dibromo-3,3′′′-dibutyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene DBrDBQT).24 The DCNDBQT was prepared by reaction of DBrDBQT with copper(I) cyanide.25
Conclusion
An endcapped thiophene oligomer, 5,5′′′-dicyano-3,3′′′-dibutyl 2,2′:5′,2′′:5′′,2′′′-quaterthiophene (DCNDBQT), was studied in its redox properties and follow-up reactions by electrochemical and spectroelectrochemical methods. Two well separated oxidation steps with the first one being reversible were found at the anodic scale, while a single quasi-reversible reduction step was detected. The cationic structure formed in the first reversible oxidation step is paramagnetic and therefore not detectable by in situ NMR spectroelectrochemistry. By lowering the temperature to 273 K the diamagnetic π-dimer of this paramagnetic structure was detected by in situ NMR experiment.In situ NMR spectroelectrochemistry shows the formation of a σ-dimer from the dication of the second oxidation step. The dimer formation is not reversible. The thiophene tetramer which is blocked by substitution in the 2- and 5-positions and sterically shielded in the 4- and 3′-positions undergoes a dimerisation in the 4′-position.
Furthermore the DCNDBQT shows a quasi-reversible reduction step. The structure formed in the reduction step is an insoluble polymer which cannot be followed by in situ NMR spectroelectrochemistry.
The reversibility of first oxidation process makes DCNDBQT useful as an hole transport material in OFFETs.23 The detailed study of dimerisation is a helpful tool for the optimisation of thin film transistor characteristics. Thus a breakdown of the device at higher potentials can be circumvented.
Acknowledgements
Financial support by the DFG-GACR grant No 203/07/JO67 is gratefully acknowledged. We thank Dr J. Tarabek (Institute of Organic and Biochemistry, CAS, Prague) for helpful discussions and F. Ziegs, M. Senf and S. Schiemenz (all of IFW Dresden) for technical assistance.Notes and references
- A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- Z. H. Li, M. S. Wong, H. Fukutani and Y. Tao, Chem. Mater., 2005, 17, 5032–5040 CrossRef CAS.
- N. Noma, T. Tsuzuki and Y. Shirota, Adv. Mater., 1995, 7, 647–648 CrossRef CAS.
- See for the different spectroscopic methods for example: (a) Spectroelectrochemistry, ed. R. J. Gale, Plenum, London and New York, 1988. Search PubMed; (b) Spectroelectrochemistry, ed, W. Kaim and A Klein, Royal Soc., London, 2008. Search PubMed; (c) IR: K. Ashley and S. Pons, Chem. Rev., 1988, 88, 673–695 Search PubMed; (d) ESR: A. Petr, L. Dunsch and A. Neudeck, J. Electroanal. Chem., 1996, 412, 153–158 Search PubMed; (e) UV-VIS: H. E. Toma and K. Araki, Curr. Org. Chem., 2002, 6, 21–34 Search PubMed; (f) Raman: L. Kavan and L. Dunsch, ChemPhysChem, 2007, 8, 975–998 Search PubMed.
- P. Rapta and L. Dunsch, J. Electroanal. Chem., 2001, 507, 287–292 CrossRef CAS.
- M. G. Hill, K. R. Mann, L. L. Miller and J.-F. Penneau, J. Am. Chem. Soc., 1992, 114, 2728–2730 CrossRef CAS; E. Levillain and J. Roncali, J. Am. Chem. Soc., 1999, 121, 8760–8765 CrossRef CAS; J.-M. Raimundo, E. Levillain, N. Gallego-Planas and J. Roncali, Electrochem. Commun., 2000, 2, 211–215 CrossRef CAS; P. Rapta, L. Kress, P. Hapiot and L. Dunsch, Phys. Chem. Chem. Phys., 2002, 4, 4181–4185 RSC.
- P. Rapta, D. Rohde, H. Hartmann and L. Dunsch, Tetrahedron Lett., 2006, 47, 7587–7590 CrossRef CAS.
- P. Rapta, O. Zeika, D. Rohde, H. Hartmann and L. Dunsch, ChemPhysChem, 2006, 7, 863–870 CrossRef CAS.
- D. Rohde, L. Dunsch, A. Tabet, H. Hartmann and J. Fabian, J. Phys. Chem. B, 2006, 110, 8223–8231 CrossRef CAS.
- M. M. M. Raposo, A. M. C. Fonseca and G. Kirsch, Tetrahedron, 2004, 60, 4071–4078 CrossRef CAS.
- Y. Liu, J. Zhou, X. Wan and Y. Chen, Tetrahedron, 2009, 65, 5209–5215 CrossRef CAS.
- R. P. Ortiz, A. Facchetti, T. J. Marks, J. Casado, M. Z. Zgierski, M. Kozaki, V. Hernandez and J. T. L. Navarrete, Adv. Funct. Mater., 2009, 19, 386–394 CrossRef CAS.
- A. Yassar, F. Demanze and D. Fichou, Opt. Mater., 1999, 12, 379–382 CrossRef CAS.
- P. Hapiot, F. Demanze, A. Yassar and F. Garnier, J. Phys. Chem., 1996, 100, 8397–8401 CrossRef CAS.
- F. Demanze, J. Cornil, F. Garnier, G. Horowitz, P. Valat, A. Yassar, R. Lazzaroni and J.-L. Bredas, J. Phys. Chem. B, 1997, 101, 4553–4558 CrossRef CAS.
- G. Zotti, S. Zecchin, B. Vercelli, A. Berlin, S. Grimoldi, M. C. Pasini and M. M. M. Raposo, Chem. Mater., 2005, 17, 6492–6502 CrossRef CAS.
- J. Casado, R. P. Ortiz, M. C. R. Delgado, R. Azumi, R. T. Oakley, V. Hernandez and J. T. L. Navarrete, J. Phys. Chem. B, 2005, 109, 10115–10125 CrossRef CAS.
- V. C. Guzman, R. P. Ortiz, M. C. R. Delgado, R. Azumi, R. T. Oakley, J. Casado, V. Hernandez and J. T. L. Navarrete, J. Mol. Struct., 2005, 744, 403–409 CrossRef.
- T. D. W. Claridge, High-Resolution NMR Techniques in Organic Chemistry, Series Editors: J.-E. Bäckvall, J. E. Baldwin and R. M. Williams, Elsevier, Amsterdam, 1999 Search PubMed.
- (a) D. W. Mincey, M. J. Popovich, P. J. Faustino, M. M. Hurst and J. A. Caruso, Anal. Chem., 1990, 62, 1197–1200 CrossRef CAS; (b) R. D. Webster, Anal. Chem., 2004, 76, 1603–1610 CrossRef CAS; (c) P. D. Prenzler, R. Bramley, S. R. Downing and G. A. Heath, Electrochem. Commun., 2000, 2, 516–521 CrossRef CAS.
- S. Klod, F. Ziegs and L. Dunsch, Anal. Chem., 2009, 81, 10262–10267 CrossRef CAS.
- K. Haubner, E. Jaehne, H. J. Adler, D. Koehler, C. Loppacher, L. Eng, J. Grenzer, A. Herasimovich and S. Scheinert, Phys. Status Solidi A, 2008, 205, 430–439 CrossRef CAS.
- T. M. Barclay, A. W. Cordes, C. D. MacKinnon, R. T. Oakley and R. Reed, Chem. Mater., 1997, 9, 981–990 CrossRef CAS.
- A. Yassar, F. Demanze, A. Jaafari, M. E. Idrissi and C. Coupry, Adv. Funct. Mater., 2002, 12, 699–708 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |