Highly enantio-, regio- and diastereo-selective one-pot [2 + 3]-cycloaddition reaction via isomerization of 3-butynoates to allenoates†, ‡
Magesh
Sampath
and
Teck-Peng
Loh
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore, 637616. E-mail: teckpeng@ntu.edu.sg; Fax: +65 6791 1961; Tel: +65 6316 8899
First published on 6th September 2010
Abstract
Phosphine-catalyzed one-pot isomerization and [2 + 3]-cycloaddition of 3-butynoates with electron-deficient olefins affords highly functionalized cyclopentenes with both good yields and excellent selectivities of up to 99%.
Highly enantio-, regio- and diastereo-selective approach towards the synthesis of functionalized five-membered rings is highly sought-after, as these are featured widely in many natural products and synthetic building blocks.1 Although, there are many methods available for the construction of five-membered rings,2 Lu's phosphine-catalyzed [2 + 3]-cycloaddition reaction using electron-deficient allenoates/2-butynoates and electron-deficient double bonds is considered to be one of the most efficient methods.3–5 This method has also been utilized in the synthesis of some natural products.6 Recently, asymmetric versions of this method have also been elegantly demonstrated by different groups.7 However, there are still limitations in the reported cases. For example, in some systems, the reported chiral phosphines are difficult to obtain and their substrate scope are relatively narrow. Another problem associated with this process is the need to use allenoates which can be difficult to make. Furthermore, the allenoates are sometime contaminated with the corresponding homo-propargylic equivalents.8 In this sense, usage of 3-butynoates which can be isomerized9 to the corresponding allenoates in situ for the [2 + 3]-cycloaddition reaction is highly desirable (Scheme 1). If successful, a mixture of propargylic and allenic esters can be directly employed in this reaction without the need for isolation.
![Proposed reaction pathways: isomerization and [2 + 3]-cycloaddition of 3-butynoates.](/image/article/2010/SC/c0sc00123f/c0sc00123f-s1.gif) | ||
| Scheme 1 Proposed reaction pathways: isomerization and [2 + 3]-cycloaddition of 3-butynoates. | ||
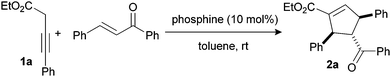
In this paper, we report the use of tributylphosphine which concurrently catalyzes the isomerization of 3-butynoates to allenoates, as well as subsequent Lu [2 + 3]-cyclization reaction of the latter. As we envisaged, in situ isomerization of 3-butynoates (1a) to allenoates led to the formation of formal [2 + 3]-cyclized product in 75% yield when trans-chalcone was stirred in a catalytic amount of tributylphosphine (Scheme 1). Notably, triphenylphosphine failed to catalyze the reaction. This is probably due to the comparatively lower nucleophilicity of triphenylphosphine as compared to aliphatic phosphines (e.g. tributylphosphine). Moreover, the reaction with triethylamine or 1,4-diazabicyclo[2.2.2]octane (DABCO) instead of tributylphosphine under the same reaction conditions, led to only isomerized product (allenoates) but not the cycloaddition or conjugate addition product.10
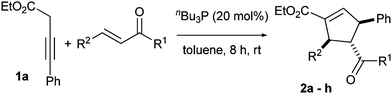
To define the generality of this method, various electron-deficient double bonds (Table 1, entries 1–8) and 3-butynoates (Table 2, entries 1–8) were screened. Both electron-withdrawing and electron-donating groups on the phenyl ring of 3-butynoates and enones furnished the products in moderate to good yields. Cycloaddition reaction with enoates such as diethyl fumarate also afforded the product (2h) in good yield (Table 1, entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Productb | Yieldc (%) |
| a See ESI1 for the detailed experimental procedure; 3-butynoates 1a contain 2% of the corresponding allenoates. b Single regio (α)- and diastereo-isomers were observed by crude NMR analysis. c Isolated yield. d Trace amount of minor diastereoisomers were observed in NMR analysis. e 10 equiv. of diethyl fumarate was used. | ||||
| 1 | C6H5 | C6H5 | 2a | 75 |
| 2 | 4-BrC6H4 | 4-FC6H4 | 2b | 72 |
| 3 | 4-MeC6H4 | 4-MeC6H4 | 2c | 77 |
| 4 | 4-ClC6H4 | 4-FC6H4 | 2d | 70 |
| 5 | 4-MeOC6H4 | 4-FC6H4 | 2e | 80 |
| 6 | C6H5 | 4-FC6H4 | 2f | 68 |
| 7d | C6H5CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H H |
C6H5 | 2g | 70 |
| 8d,e | OEt | COOEt | 2h | 78 |
|
|
|||
|---|---|---|---|
| Entry | R | Productb | Yieldc (%) |
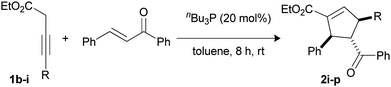
| a See the ESI for the detailed experimental procedure; 3-butynoate 1e was contaminated with 35% of the corresponding allenoate and the remainder of the 3-butynoates contain 5% of the allenoates. b Single regio (α)- and diastereo-isomers were observed by crude NMR analysis. c Isolated yield. d Reaction required 28 h. e Product 2o was hydrolyzed to the corresponding acid (Scheme 2) and the relative stereochemistry was confirmed by X-ray analysis (CCDC 748921). f Corresponding allenoates were used. Compound 1i was synthesized using the procedure reported in the literature.11 | |||
| 1 | 4-MeC6H41b | 2i | 77 |
| 2 | 4-MeOC6H41c | 2j | 82 |
| 3 | (2-Me)(4-MeO)C6H31d | 2k | 88 |
| 4 | 4-CF3C6H41e | 2l | 71 |
| 5 | 3-Thienyl 1f | 2m | 78 |
| 6 | 6-MeO-2-naphthyl 1g | 2n | 80 |
| 7d,e | Cyclopropyl 1h | 2o | 85 |
| 8f | CH31i | 2p | 87 |
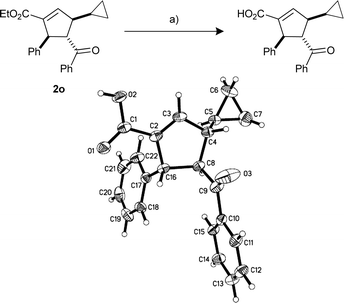 | ||
Scheme 2 Hydrolysis of product 2o; X-Ray crystallographic structure of hydrolyzed product; 50% probability was chosen for the ellipsoids. Reagents and conditions: (a) LiOH·H2O (5 equiv.), THF–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 60 °C, 8 h, 95%. :1), 60 °C, 8 h, 95%. | ||
In addition, 3-butynoates bearing thiophene (1f), naphthalene (1g) and aliphatic substituents such as the cyclopropyl group (1h) also afforded the products in good yields (Table 2, entries 5–7).
Next, we focused on the asymmetric version of this reaction. On the basis of our studies as described above, we believed that aliphatic phosphines with stereogenic centers in close proximity to the reactive sites will be more promising for asymmetric cycloaddition reaction. With this in mind, various commercially available chiral phosphines were screened using a solution of 3-butynoate (1a) (0.26 mmol), trans-chalcone (0.29 mmol) and 10 mol% of phosphines in dry toluene (1.5 mL) at room temperature (Table 3, entries 1–8).
|
|
||||
|---|---|---|---|---|
| Entry | Phosphinea | t/h | Yieldb (%) | Eec (%) |
| a See Scheme 3 for the structure of commercially available chiral phosphines screened. b Isolated yield. c Ee (%) was determined using chiral HPLC. | ||||
| 1 | (+)-DIOP | 8 | 72 | 66 |
| 2 | (R,R)-Et-BPE | 8 | 78 | 63 |
| 3 | (R,R)-DIPAMP | 8 | 87 | 95 |
| 4 | (R,R)-Et-DUPHOS | 24 | 27 | 33 |
| 5 | (S)-(−)-2-[2-(Diphenylphosphino)phenyl]-4-isopropyl-2-oxazoline | 24 | — | — |
| 6 | (R)-BINAP | 24 | — | — |
| 7 | (R)-Tol-BINAP | 24 | — | — |
| 8 | (2S,3S)-CHIRAPHOS | 24 | 58 | 34 |
![Structure of commercially available chiral phosphines screened for asymmetric [2 + 3]-cycloaddition reaction.](/image/article/2010/SC/c0sc00123f/c0sc00123f-s3.gif) | ||
| Scheme 3 Structure of commercially available chiral phosphines screened for asymmetric [2 + 3]-cycloaddition reaction. | ||
The commercially available catalyst (R,R)-DIPAMP emerged as the best catalyst in terms of both yield and enantioselectivity. However, lower yield and enantioselectivity were observed when the catalyst loading was decreased to 5 mol%. Increasing the amount of catalyst loading to 20 mol% and decreasing the temperature of the reaction to 0 °C did not increase the yield or enantioselectivity of the product. Using the best chiral phosphine (R,R)-DIPAMP and optimized reaction conditions, asymmetric reactions were carried out with a series of electron-deficient enones (Table 4, entries 1–8). In all the cases, both excellent enantioselectivities and yields were observed. Notably, reaction with symmetrical dienone affords single [2 + 3]-cycloaddition product (2g′) with excellent enantioselectivity (Table 4, entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product/yieldc (%) | Eed (%) |
| a See ESI1 for the detailed experimental procedure. b Single regio (α)- and diastereo-isomers were observed by crude NMR analysis. c Isolated yield. d Ee was determined using chiral HPLC (see ESI1 for more details). e Symmetrical dienone was used. No trace of double [2 + 3]-cyclized product was observed. f 10 equiv. of diethyl fumarate was used. g Reaction completed in 12 h. | ||||
| 1 | C6H5 | C6H5 | 2a′/87 | 95 |
| 2 | 4-BrC6H4 | 4-FC6H4 | 2b′/92 | 95 |
| 3 | 4-MeC6H4 | 4-MeC6H4 | 2c′/95 | 93 |
| 4 | 4-ClC6H4 | 4-FC6H4 | 2d′/88 | 95 |
| 5 | 4-MeOC6H4 | 4-FC6H4 | 2e′/82 | 98 |
| 6 | C6H5 | 4-FC6H4 | 2f′/85 | 95 |
| 7e | C6H5CHCH |
C6H5 | 2g′/90 | 99 |
| 8f,g | OEt | COOEt | 2h′/88 | 81 |
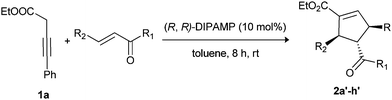
To further explore the generality of this catalyst DIPAMP, electronically and sterically divergent 3-butynoates were screened (Table 5, entries 1–8). In all the cases, excellent yields and enantioselectivities were obtained. Interestingly, the catalyst (R,R)-DIPAMP afforded excellent enantioselectivities with butynoates containing thiophene (1f) (Table 5, entry 5) and naphthalene (1g) (Table 5, entry 6). However, (R,R)-DIPAMP failed to yield the desired product with cyclopropyl substituted 3-butynoate (1h) even after stirring for 3 days. It was gratifying to find that addition of a catalytic amount 10 mol% of triethylamine facilitated the isomerization to afford the cyclized product in good yield with high enantioselectivity (Table 5, entry 7). Interestingly, in all the cases, products were obtained as single regio- and diastereo-isomers, which was consistent with the results reported by Miller and Cowen.7c
|
|
|||
|---|---|---|---|
| Entry | R | Product/yieldc (%) | Eed (%) |
| a See ESI1 for the detailed experimental procedure. b Single regio (α)- and diastereo-isomers were observed by crude NMR analysis. c Isolated yield. d Ee was determined using chiral HPLC (see ESI1 for more details). e 3-Butynoate 1e contaminated with 35% of the corresponding allenoate and the remainder of the 3-butynoates contain 5% of allenoates. f 10 mol% of triethylamine was added and the reaction was completed in 12 h. g Absolute configuration was assigned by comparing with the optical rotation value reported in the literature.7c h Corresponding allenoate was used (for the synthesis of allenoate see ref. 11). | |||
| 1 | 4-MeC6H41b | 2i′/82 | 97 |
| 2 | 4-MeOC6H41c | 2j′/89 | 95 |
| 3 | (2-Me)(4-OMe)C6H31d | 2k′/93 | 84 |
| 4e | 4-CF3C6H41e | 2l′/66 | 94 |
| 5 | 3-Thienyl 1f | 2m′/90 | 96 |
| 6 | 6-OMe-2-naphthyl 1g | 2n′/77 | 96 |
| 7f | Cyclopropyl 1h | 2o′/93 | 90 |
| 8g,h | CH31i | 2p′/87 | 99 |
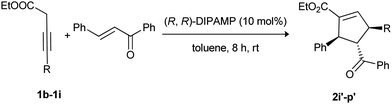
The absolute configuration of the product 2p′ was assigned by comparing the optical rotation of the corresponding benzyl ester with literature value.7c The product (2p′) was transformed to the corresponding benzyl ester as described in Scheme 4.
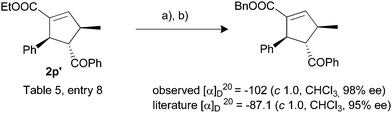 | ||
| Scheme 4 Determination of absolute stereochemistry; see ESI† for detailed experimental procedure. Reagents and conditions: (a) LiOH·H2O (5 equiv.), THF–H2O (1:1), 60 °C, 5 h, 92%; (b) PhCH2Br, K2CO3, DMF, rt, 12 h, 95%. | ||
A control experiment had been carried out by first isolating the (±)-allenoate intermediate before subjecting it to the asymmetric [2 + 3]-cycloaddition using trans-chalcone and 10 mol% of (R,R)-DIPAMP (Scheme 5).12 The above reaction affords identical results with the one-pot [2 + 3]-cycloaddition of 3-butynoates using trans-chalcone and (R,R)-DIPAMP (Table 4, entry 1). This experiment indicated that the chirality of the allenoates played no significant role in the asymmetric induction of the reaction.
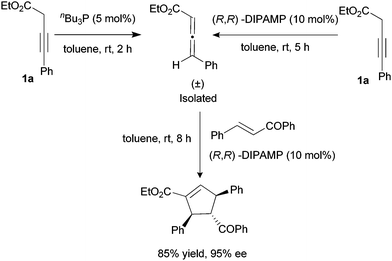 | ||
| Scheme 5 A control experiment: isolation of intermediate. | ||
In conclusion, we have demonstrated the direct applicability of 3-butynoates in the [2 + 3]-cycloaddition reaction. This one-pot procedure is an attractive alternative to the usual protocol reported for this type of reaction. In addition, we have identified a more efficient commercially available chiral phosphine, (R,R)-DIPAMP for this cycloaddition reaction, affording various cyclopentene derivatives in high optical purities. Further investigation of the scope, mechanism and application of this methodology to the synthesis of complex molecules are in progress.
Acknowledgements
We thank Dr Yong-Xin Li for X-ray analyses. We gratefully acknowledge the Nanyang Technological University and Biomedical Research Council (A*STAR Grant No 05/1/22/19/408) for the funding support of this researchNotes and references
- (a) R. C. Hartley and S. T. Caldwell, J. Chem. Soc., Perkin Trans. 1, 2000, 477–501 RSC; (b) H. Wu, H. Zhang and G. Zhao, Tetrahedron, 2007, 63, 6454–6461 CrossRef CAS.
- For reviews, see: (a) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102–3116 RSC , and references therein; (b) T. Hudlicky and J. D. Price, Chem. Rev., 1989, 89, 1467–1486 CrossRef CAS; (c) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49–92 CrossRef CAS; (d) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1986, 25, 1–20 CrossRef; (e) H. Li and T. P. Loh, J. Am. Chem. Soc., 2008, 130, 7194–7195 CrossRef; (f) S. G. Van Ornum and J. M. Cook, Tetrahedron Lett., 1997, 38, 3657–3658 CrossRef CAS; (g) B. M. Trost, P. Seoane, S. Mignani and M. Acemoglu, J. Am. Chem. Soc., 1989, 111, 7487–7500 CrossRef CAS; (h) A. Panossian, N. F. Bregeot and A. Marinetti, Eur. J. Org. Chem., 2008, 3826–3833.
- (a) X. Lu and C. Zhang, J. Org. Chem., 1995, 60, 2906–2908 CrossRef CAS. For phosphine catalyzed annulation reactions, see: (b) S. Zheng and X. Lu, Org. Lett., 2009, 11, 3978–3981 CrossRef CAS; (c) Z. Lu, S. Zheng, X. Zhang and X. Lu, Org. Lett., 2008, 10, 3267–3270 CrossRef; (d) M. Sampath and T. P. Loh, Chem. Commun., 2009, 1568–1570 RSC; (e) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS; (f) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140–1152 RSC; (g) L. Jean and A. Marinetti, Tetrahedron Lett., 2006, 47, 2141–2145 CrossRef CAS; (h) Y. Du, X. Lu and Y. Yu, J. Org. Chem., 2002, 67, 8901–8905 CrossRef CAS; (i) Y. S. Tran and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12632–12633 CrossRef CAS; (j) X. F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716–4717 CrossRef CAS; (k) G. S. Creech and O. Kwon, Org. Lett., 2008, 10, 429–432 CrossRef CAS; (l) X. F. Zhu, C. E. Henry, J. Wang, T. Dudding and O. Kwon, Org. Lett., 2005, 7, 1387–1390 CrossRef CAS; (m) X. F. Zhu, A. P. Schaffner, R. C. Li and O. Kwon, Org. Lett., 2005, 7, 2977–2980 CrossRef CAS; (n) C. E. Henry and O. Kwon, Org. Lett., 2007, 9, 3069–3072 CrossRef CAS; (o) Z. Xu and X. Lu, Tetrahedron Lett., 1999, 40, 549–552 CrossRef CAS.
- (a) Y. S. Tran and O. Kwon, Org. Lett., 2005, 7, 4289–4291 CrossRef CAS; (b) V. Sriramurthy, G. A. Barcan and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12928–12929 CrossRef CAS; (c) R. P. Wurz and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 12234–12235 CrossRef CAS; (d) A. T. Ung, K. Schafer, K. B. Lindsay, S. G. Pyne, K. Amornraksa, R. Wouters, I. V. Linden, I. Biesmans, A. S. J. Lesage, B. W. Skelton and A. H. White, J. Org. Chem., 2002, 67, 227–233 CrossRef CAS; (e) A. Scherer and J. A. Gladysz, Tetrahedron Lett., 2006, 47, 6335–6337 CrossRef CAS; (f) X. Meng, Y. Huang and R. Chen, Org. Lett., 2009, 11, 137–140 CrossRef CAS; (g) H. Guo, Q. Xu and O. Kwon, J. Am. Chem. Soc., 2009, 131, 6318–6319 CrossRef CAS; (h) X. Meng, Y. Huang, H. Zhao, P. Xie, J. Ma and R. Chen, Org. Lett., 2009, 11, 991–994 CrossRef CAS; (i) X.-Y. Guan and M. Shi, J. Org. Chem., 2009, 74, 1977–1981 CrossRef CAS; (j) V. Nair, A. T. Biju, K. Mohanan and E. Suresh, Org. Lett., 2006, 8, 2213–2216 CrossRef CAS; (k) D. J. Wallace, R. L. Sidda and R. A. Reamer, J. Org. Chem., 2007, 72, 1051–1054 CrossRef CAS; (l) L.-W. Ye, X.-L. Sun, Q.-G. Wang and Y. Tong, Angew. Chem., Int. Ed., 2007, 46, 5951–5954 CrossRef CAS.
- For mechanistic investigation of [3 + 2]-cycloaddition reactions, see: (a) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470–3471 CrossRef CAS; (b) Y. Liang, S. Liu and Z.-X. Yu, Synlett, 2009, 905–909 CAS; (c) T. Dudding, O. Kwon and E. Mercier, Org. Lett., 2006, 8, 3643–3646 CrossRef CAS; (d) E. Mercier, B. Fonovic, C. Henry, O. Kwon and T. Dudding, Tetrahedron Lett., 2007, 48, 3617–3620 CrossRef CAS.
- (a) Y. Du and X. Lu, J. Org. Chem., 2003, 68, 6463–6465 CrossRef CAS; (b) J. C. Wang and M. J. Krische, Angew. Chem., Int. Ed., 2003, 42, 5855–5857 CrossRef CAS; (c) T. Q. Pham, S. G. Pyne, B. W. Skelton and A. H. White, J. Org. Chem., 2005, 70, 6369–6377 CrossRef CAS; (d) H. Mizuno, K. Domon, K. Masuya, K. Tanino and I. Kuwajima, J. Org. Chem., 1999, 64, 2648–2656 CrossRef CAS.
- (a) G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Am. Chem. Soc., 1997, 119, 3836–3837 CrossRef CAS; (b) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426–1429 CrossRef CAS; (c) B. J. Cowen and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 10988–10989 CrossRef CAS; (d) Y. Q. Fang and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 5660–5661 CrossRef CAS; (e) A. Voituriez, A. Panossian, N. F. Bregeot, P. Retailleau and A. Marinetti, J. Am. Chem. Soc., 2008, 130, 14030–14031 CrossRef; (f) A. Voituriez, A. Panossian, N. F. Bregeot, P. Retailleau and A. Marinetti, Adv. Synth. Catal., 2009, 351, 1968–1976 CrossRef CAS.
- (a) M. J. Lin and T. P. Loh, J. Am. Chem. Soc., 2003, 125, 13042–13043 CrossRef; (b) F. Fu, L. M. Hoang and T. P. Loh, Org. Lett., 2008, 10, 3437–3439 CrossRef; (c) F. Sato, T. Nakagawa and A. Kasatkin, Tetrahedron Lett., 1995, 36, 3207–3210 CrossRef CAS; (d) U. Maeorg and A. Jogi, Molecules, 2001, 6, 964–968 Search PubMed; (e) W. L. Wu, Z. J. Yao, Y. L. Li, J. C. Li, Y. Xia and Y. L. Wu, J. Org. Chem., 1995, 60, 3257–3259 CrossRef CAS; (f) M. Iyoda, Y. Kanao, M. Nishizaki and M. Oda, Bull. Chem. Soc. Jpn., 1989, 62, 3380–3382 CAS; (g) A. Suárez and G. C. Fu, Angew. Chem., Int. Ed., 2004, 43, 3580–3582 CrossRef CAS; (h) K. Takami, S. Usugi, H. Yorimitsu and K. Oshima, Synthesis, 2005, 824–839 CAS; (i) J. A. Cabezas and L. X. Alvarez, Tetrahedron Lett., 1998, 39, 3935–3938 CrossRef CAS; (j) W. Miao and T. H. Chan, Synthesis, 2003, 785–789 CAS.
- For base-promoted isomerization, see: (a) M. E. Jung, M. Node, R. W. Pfluger, M. A. Lyster and J. A. Lowe III, J. Org. Chem., 1982, 47, 1150–1152 CrossRef CAS; (b) D. Ma, Y. Yu and X. Lu, J. Org. Chem., 1989, 54, 1105–1109 CrossRef CAS; (c) H. Liu, D. Leow, K. W. Huang and C. H. Tan, J. Am. Chem. Soc., 2009, 131, 7212–7213 CrossRef; (d) M. Oku, S. Arai, K. Katayama and T. Shioiri, Synlett, 2000, 493–494 CAS.
- C. A. Evans and S. J. Miller, J. Am. Chem. Soc., 2003, 125, 12394–12395 CrossRef CAS.
- R. W. Lang and H. J. Hansen, Org. Synth., 1984, 62, 202 CAS.
- For control experiment see ESI†.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical data. CCDC reference number 748921. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00123f |
| ‡ General procedure for the synthesis of 2a: To a stirred solution of 3-butynoate 1a (50 mg; 0.265 mmol) and trans-chalcone (61 mg, 0.292 mmol) in toluene (1.5 mL) was added (R,R-DIPAMP) (12 mg, 0.026 mmol; pre-dissolved in toluene) dropwise at 0 °C under nitrogen. After 8 h stirring at room temperature under N2 atmosphere, the reaction mixture was concentrated and purified using flash column chromatography (10% ethyl acetate in hexane) to afford pure product 2a (89.5 mg, 87% yield, 95% ee). |
| This journal is © The Royal Society of Chemistry 2010 |