Heterolytic and heterotopic dissociation of hydrogen on ceria-supported gold nanoparticles. Combined inelastic neutron scattering and FT-IR spectroscopic study on the nature and reactivity of surface hydrogen species†
Raquel
Juárez
a,
Stewart F.
Parker
b,
Patricia
Concepción
a,
Avelino
Corma
a and
Hermenegildo
García
*a
aInstituto Universitario de Tecnología Química CSIC-UPV, Universidad Politécnica de Valencia, 46022, Valencia, Spain. E-mail: hgarcia@qim.upv.es
bISIS Facility, STFC Rutherford Appleton Laboratory, Chilton, Didcot, Oxon, UK OX11 0QX
First published on 27th August 2010
Abstract
Inelastic neutron scattering of gold nanoparticles (3–4 nm, 0.48 wt%) supported on nanoparticulate ceria (5 nm) (Au/CeO2) shows that hydrogen treatment of Au/CeO2 gives rise to the appearance of a new band (400–600 cm−1) corresponding to bridging hydroxyl groups. This band disappears (without observing the appearance of any new band) when the sample is exposed to CO2 or O2. FT-IR spectroscopy on the same samples shows the formation of Au–H after hydrogen chemisorption. The Au–H band also disappears when the sample is subsequently treated with CO2 or O2. In the last case formation of hydroperoxide is observed. The band assignments were supported by isotopic labelling with D2 and 18O2. The above data clearly establish that upon treatment of Au/CeO2 with hydrogen at 150 °C, heterolytic bond cleavage of dihydrogen occurs leading to a proton bonded to a ceria oxygen and a hydride bonded to gold.
Introduction
Catalysis by supported gold nanoparticles is an area of much current interest mainly because it constitutes a remarkable example of the influence of the nanometre length scale on the properties of the resulting materials.1–6 Since the pioneering work of Haruta, it has become clear that the catalytic activity of gold nanoparticles decreases as the particle size increases, the activity disappearing for particles larger than 20 nm.7,8 Thus, in contrast to the high catalytic activity of other noble metals, gold only becomes catalytically active when the particle size is below a few nanometres.From the extensive work carried out in this area it can be deduced that gold nanoparticles are more general catalysts for oxidations than for reductions.9 This is a remarkable feature of Au compared to Pt, Rh, Ru and other noble metals that besides oxidation are also excellent hydrogenation catalysts.10–12 Nevertheless, the lower activity of Au to promote hydrogenation of multiple C–C bonds has recently resulted in a new selective hydrogenation of nitro groups in the presence of other reducible functional groups and particularly in the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.13–15 The relatively poor activity of Au for hydrogenation of C–C multiple bonds has been attributed in part to the inability of Au to easily break the H–H bond and to the instability of Au hydrides.4,16,17 Compared to Pd and Pt, Au does not form stable hydrides under the conditions in which Pd and Pt do form hydrides.18 It has been recently shown that a small percentage of Pt decorating Au nanoparticles remarkably increases the catalytic activity of supported gold nanoparticles due to the high activity of Pt to break the H–H bond, producing hydrogen spillover.19
C bonds.13–15 The relatively poor activity of Au for hydrogenation of C–C multiple bonds has been attributed in part to the inability of Au to easily break the H–H bond and to the instability of Au hydrides.4,16,17 Compared to Pd and Pt, Au does not form stable hydrides under the conditions in which Pd and Pt do form hydrides.18 It has been recently shown that a small percentage of Pt decorating Au nanoparticles remarkably increases the catalytic activity of supported gold nanoparticles due to the high activity of Pt to break the H–H bond, producing hydrogen spillover.19
In spite of the general difficulty to generate Au hydrides and the instability of these species, there are precedents in the literature in which Au hydride has been characterized in the gas phase as well as in matrices at low temperature.20–22 Also, dimeric Au hydride, AuCl[xantphos] and AuCl[xy-xantphos] [xantphos: 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene] complexes have been characterized by liquid 1H and 3P NMR spectroscopy as reaction intermediates in the hydrogenative silylation of alcohols.23,24 For supported gold catalysts, theoretical calculations suggest that dihydrogen on gold becomes dissociated preferentially on low coordination corner or edge gold atoms not directly bonded to O atoms of the support or forming the first layer in contact with the support.13,25,26
In view of these precedents, it is of interest to carry out an experimental study of the species generated upon chemisorption of molecular hydrogen on supported Au catalysts. One case of these gold catalysts of particular interest is gold supported on nanoparticulate ceria (Au/CeO2), because this material has been found to exhibit high catalytic activity for many oxidation and hydrogenation reactions.27–29 Moreover, for this material a synergy or cooperation of gold and ceria nanoparticles has been proposed.30 Herein, we will show that the presence of gold nanoparticles on the surface of ceria dramatically changes the interaction of ceria with hydrogen, producing heterolytic hydrogen dissociation. There are precedents in the literature showing the heterolytic splitting of H2 on metal oxides, such as gallium oxide,31 but in the present case we will show that gold nanoparticles play a key role in promoting the dissociation.
In the present work we have carried out an in situ spectroscopic study of the hydrogen species present on Au/CeO2 combining inelastic neutron scattering (INS) spectroscopy and IR spectroscopy. The advantages of the combined study are the high selectivity of INS to monitor hydrogen species and the higher sensitivity of IR spectroscopy. After chemisorption of hydrogen on the surface of Au/CeO2, the reactivity of the surface hydrogen species for CO2 hydrogenation and for the formation of hydrogen peroxide by reaction with dioxygen has been studied. The results are compatible with the heterolytic dissociation of H2 on the surface of Au/CeO2, leading to the formation of Au hydrides as well as bridging hydroxyl groups on the ceria (“heterolytic and heterotopic dissociation”). In one scarce precedent using INS for the study of a gold catalyst, Goodman and co-workers detected the presence of hydroperoxy species on the surface of Au/TiO2 by using this material as a catalyst for the reaction of H2 and O2 at 150 °C and sudden quenching of the catalyst under liquid nitrogen.32 However, this approach did not allow detection of the primary hydrogen species arising from chemisorption of hydrogen on the gold and, moreover, it does not correspond to the Au/CeO2 catalyst. We will show that Au/CeO2 exhibits a distinctive hydrogen chemisorption activity that is not found in Au/TiO2 and, therefore, these two gold catalysts are hardly comparable.
Results and discussion
Catalysts
The samples under study were nanoparticulate CeO2 (5 nm average particle size) prepared by controlled hydrolysis of Ce3+ in aqueous solution and Au/CeO2 prepared by the conventional deposition-precipitation method with a gold loading of 0.48 wt%.30,33,34 This small gold loading is known to render highly active Au/CeO2 catalysts comprised of homogeneously dispersed gold nanoparticles of narrow size distribution (2–4 nm diameter) on the surface of CeO2 nanoparticles.33,34 The samples were characterized by TEM that showed images in agreement with those previously reported for these types of samples. The reference Au/TiO2 is a commercial sample supplied by the World Gold Council that contains 1.5 wt% of Au present as small metal nanoparticles (3–4 nm) supported on P-25 titania nanoparticles.Hydrogen chemisorption
Preliminary temperature programmed reduction (TPR) measurements revealed remarkable differences between the profiles of hydrogen uptake for CeO2 and Au/CeO2. Fig. 1 shows the hydrogen uptake profiles for CeO2, Au/CeO2 and Au/TiO2. The reference Au/TiO2 sample used here is analogous to the Au/TiO2 sample previously studied by Goodman et al.32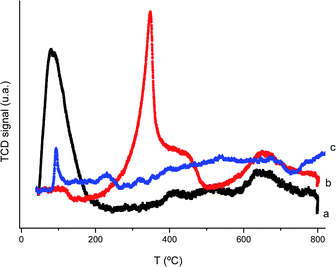 | ||
| Fig. 1 Thermoprogrammed hydrogen reduction of Au/CeO2 (a, black line), CeO2 (b, red line), and Au/TiO2 (c, blue line). | ||
As it can be seen in Fig. 1, CeO2 exhibits two chemisorption peaks having maxima at 373 and 656 °C. Upon deposition of such a small percentage of 0.48 wt% of gold on ceria, remarkable changes in the hydrogen that reacts/adsorbs on the solid occur. The hydrogen chemisorption peak at 373 °C disappears and is replaced by a double peak of approximately the same area with maxima at 63 and 95 °C. This low temperature hydrogen chemisorption is characteristic of supported gold samples and is also present, although considerably less intense, in Au/TiO2 (plot c in Fig. 1). This low temperature hydrogen chemisorption is typically attributed to the reduction of positive gold atoms remaining on the solid support or to hydrogen spillover on the solid. The high intensity of the low temperature peak of hydrogen uptake in Au/CeO2 accompanied by the disappearance of the 373 °C peak of the ceria agrees more with the occurrence of two different processes, i.e., reduction of ceria framework oxygens forming water and hydrogen splitting. For this reason, the amount of hydrogen chemisorbed is higher than the amount that will correspond to the exclusive reduction of Au atoms, particularly considering the low Au loading. This indicates that a small proportion of Au is sufficient to produce remarkable effects in the reducibility of the material and in the catalytic activity.27,35 This suggests that hydrogen reacts with ceria framework oxygens, besides becoming chemisorbed on the support. The high-temperature reduction peak in both CeO2 and Au/CeO2 undergoes a relatively minor shift from 656 to 624 °C.
In contrast to Au/CeO2, the hydrogen uptake plot of Au/TiO2 only exhibits a weak hydrogen uptake below 100 °C, indicating that hydrogen spillover does not occur on the surface of P25 titania and showing that the reducibility of Au/CeO2 is unique among gold catalysts. This can be interpreted by considering that upon particle size decrease on the nanometre scale, ceria undergoes structural changes consisting of the creation of oxygen vacancies and partial Ce(IV) to Ce(III) reduction. One remarkable consequence of the particle size reduction on ceria is the change in its properties from insulator to semiconductor.36 Apparently, the presence of gold in minor amounts increases the formation of structural defects on ceria and the presence of these defects is reflected in the hydrogen reaction (with ceria framework oxygens) and chemisorption and also in the catalytic properties of nanoparticulate ceria.34
INS spectra of chemisorbed hydrogen on Au/CeO2
The TPR experiments presented here show that Au/CeO2 chemisorbs a significant amount of hydrogen at relatively low temperature. This is quite surprising considering that as noted earlier Au/CeO2 does not exhibit a high catalytic activity for hydrogenation reactions of C–C multiple bonds. We were interested in determining how hydrogen is adsorbed, which species are generated as consequence of this adsorption and their reactivity with simple probe molecules. In order to determine the fate of hydrogen chemisorbed on Au/CeO2 and CeO2 samples we performed an INS study of Au/CeO2 and CeO2 before and after adsorption of hydrogen. Fig. 2 and 3 show the accessible energy region of the series of INS spectra recorded for CeO2 and Au/CeO2, respectively.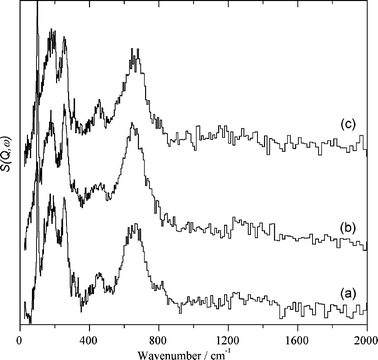 | ||
| Fig. 2 INS spectra recorded at 20 K for nanoparticulate CeO2 after outgassing this sample at 150 °C under reduced pressure for 1 h (a) and after dosing the sample with hydrogen at 150 °C for 1 h and subsequent outgassing under reduced pressure at 150 °C for 1 h (b). Spectrum c was recorded at 20 K for the sample corresponding to spectrum b after dosing with CO2 at 150 °C for 1 h followed by 1 h evacuation at 150 °C under reduced pressure. (The intense features in the 0–400 cm−1 region are due to the Inconel sample can). | ||
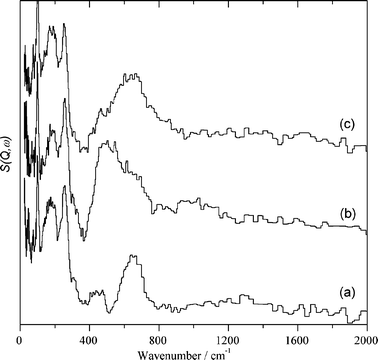 | ||
| Fig. 3 INS spectra recorded at 20 K for the Au/CeO2 sample after outgassing at 150 °C for 1 h under vacuum (a), subsequent exposure to H2 at 150 °C for 1 h (b), and exposing the hydrogen-dosed Au/CeO2 to CO2 at 150 °C for 1 h (c). | ||
After outgassing CeO2 at 150 °C and subsequent exposure of the outgassed sample to hydrogen at 150 °C some hydrogen uptake was monitored for this sample. Table 1 lists the amount of hydrogen adsorbed on CeO2 in the INS experiment. The INS spectra for CeO2 before and after exposure to hydrogen shown in Fig. 2 (spectra “a” and “b”) were coincident and INS spectroscopy was insensitive to the hydrogen uptake. Only the presence of terminal hydroxyl groups was detected in the sample, as indicated by the presence of a band at 662 cm−1 accompanied by other vibration peaks about 100 cm−1. The difference spectra between the spectra of the sample after and before dosing with hydrogen only reveals a weak broad absorption band from 200 to 1000 cm−1 attributable to the presence of adsorbed water on the solid surface of nanoparticulate ceria formed during the hydrogen adsorption.
| Catalyst | Uptake | |||
|---|---|---|---|---|
| H2/mmol (g cat)−1 | CO2/mmol (g cat)−1 | H2/mmol (g cat)−1 | O2/mmol (g cat)−1 | |
| a Adsorption conditions: first the sample (20 or 19 g for CeO2 or Au/CeO2, respectively) placed in a can is outgassed at 150 °C under vacuum (10−5 Torr) for 1 h. After equilibrating at room temperature, the sample is exposed to H2 and the INS spectrum recorded. Subsequently, the sample is exposed to CO2 or O2 and heated at 150 °C for 1 h. The two H2 columns refer to the H2 adsorbed before the CO2 or O2 measurement, respectively. | ||||
| CeO2 | 0.23 | 0.52 | 0.23 | <0.01 |
| Au/CeO2 | 0.63 | 0.74 | 0.42 | 0.23 |
In spite of the low hydrogen uptake (see Table 1) we submitted the CeO2 sample dosed with hydrogen to subsequent treatments with CO2 and O2, because they constitute valid control measurements to compare the catalytic behaviour of CeO2 and Au/CeO2. It was noted that the hydrogen-treated CeO2 sample did not adsorb O2, but adsorbed a significant amount of CO2. Table 1 contains the O2 and CO2 uptake of the CeO2 sample under study when submitted to the thermal treatments at 150 °C. As expected in view of the lack of O2 adsorption, no changes in the INS spectra were observed for CeO2 when the hydrogen-dosed sample was subsequently treated with oxygen. Likewise, and in spite of the large amount of CO2 adsorbed on CeO2, Fig. 2 spectrum “c” shows that no changes in the INS spectrum of the hydrogen-dosed CeO2 sample were observed when exposed to CO2. This indicates that the terminal hydroxyl groups of CeO2 are unreactive towards CO2. This point is catalytically relevant since it has been determined that, in contrast to supported gold catalysts, CeO2 does not promote hydrogenation of CO2, but it can act as a catalyst for CO2 insertions.37,38 These observations are again in agreement with the lack of hydrogen chemisorption on the surface of CeO2. We will come back to the species formed upon CO2 adsorption on CeO2 when presenting the results of the IR spectroscopy study (see below).
Similar experiments to those previously described for CeO2 were carried out with Au/CeO2 and Au/TiO2 that are the actual catalysts for hydrogenations, oxidations and other reactions.9,30,33 No significant hydrogen uptake was observed for Au/TiO2 and accordingly INS spectroscopy failed to detect on Au/TiO2 any species derived from hydrogen adsorption. In contrast to this, the spectra recorded for Au/CeO2 after outgassing at 150 °C and subsequent treatments with H2 followed by treatment of the hydrogen-dosed Au/CeO2 to CO2 and O2 exhibit significant variations. Fig. 3, 4 and 5 present the INS spectra recorded for Au/CeO2 under the various treatments.
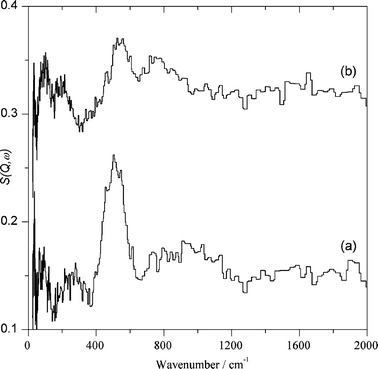 | ||
| Fig. 4 Difference spectra obtained for Au/CeO2 by subtracting the spectra shown in Fig. 3: (a) after adsorbing H2 at 150 °C with the background subtracted; (b) after adsorbing CO2 at 150 °C with the background subtracted (the spectra are plotted on the same scale but with (b) displaced by 0.15 vertically for clarity). | ||
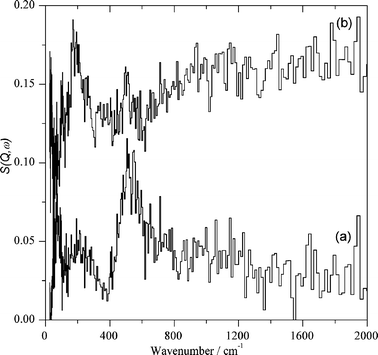 | ||
| Fig. 5 Difference INS spectra obtained for Au/CeO2: (a) after adsorbing H2 at 150 °C with the background subtracted; (b) sample corresponding to spectrum a after absorbing oxygen. | ||
As it can be seen in Fig. 3, after outgassing at 150 °C under vacuum Au/CeO2 shows the band appearing at 662 cm−1 accompanied by peaks below 100 cm−1 that are coincident with those previously commented on for the CeO2 support and can be attributed to terminal hydroxyl groups of the CeO2 support. However, we also noticed a much less intense peak from 400 to 500 cm−1. This low intensity peak was absent in the CeO2 support and can be attributed to hydroxyl groups of the CeO2 modified by the interaction of the support with gold nanoparticles.
When this outgassed Au/CeO2 sample was exposed to hydrogen gas at 150 °C, significant hydrogen uptake was measured. Table 1 also contains data regarding the amount of hydrogen chemisorbed by Au/CeO2 under the experimental conditions used. The INS spectrum of the Au/CeO2 solid dosed with hydrogen is also shown in Fig. 3. As can be seen there, after hydrogen chemisorption the two resolved bands present in evacuated Au/CeO2 appear as a broad envelope extending from 400 to 750 cm−1. Also the spectral baseline from 750 to 1300 cm−1 grows, although the intensity in this region is less than that of the 400 to 750 cm−1 region. Importantly, perusal of spectra b in Fig. 2 and 3 clearly provides spectroscopic evidence of the different chemisorption properties of CeO2 and Au/CeO2.
Considerably more informative than the INS spectra are the difference spectra. Fig. 4 shows the set of difference spectra. As can be seen, the difference INS spectrum after and before hydrogen exposure clearly shows the new bands formed by hydrogen chemisorption on the dehydrated Au/CeO2 sample appearing from 400 to 600 cm−1 accompanied by a broad band from 750 to 1200 cm−1. This second band is attributed to physisorbed solid water present on the catalyst surface at the temperature in which the INS measurements are performed (20 K). Concerning the assignment of the 400–600 cm−1 intense peak, we attributed it to bridging hydroxyl groups due to its wavenumber and the fact that terminal hydroxyl groups should exhibit an additional vibration band around 100 cm−1 accompanying the main peak and that this additional band is not observed.
It is noteworthy that in the INS measurements the presence of water cannot be considered as due to contamination since analogous experiments with control samples conclusively rule out this possibility. Thus, it can be assumed that some lattice oxygen of nanoparticulate ceria reacts with hydrogen to form the water observed.
Assignment of the 400–600 cm−1 band to weakly-bound, physisorbed hydrogen interacting with the solid surface can be ruled out by the absence of the characteristic J 0 ← 1 rotational transition at ∼120 cm−1. The spectral changes shown in Fig. 4 arising due to hydrogen chemisorption were reproducible, including the amount of gas uptake and peak positions and intensity.
Based on the observed INS spectra, the following processes indicated in eqn (1) and (2) can be proposed for hydrogen chemisorption on Au/CeO2. This proposal is compatible with the FTIR spectra of the same samples discussed below (particularly with the observation of the Au–H band) and with the bridging nature of the hydroxyl group observed in INS. Apparently, the lower sensitivity inherent to INS spectroscopy and the low percentage of Au on the material are responsible for this lack of observation of the Au–H in INS.
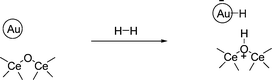 | (1) |
| 2CeO2 + H2 → Ce2O3 + H2O | (2) |
Reaction of hydrogen surface species with CO2 and O2 monitored by INS
It has been reported that supported gold is a hydrogenation catalyst for CO2.39–41 We wanted to determine the reactivity of the surface hydrogen species with CO2. Exposure of Au/CeO2 previously dosed with hydrogen to CO2 at 150 °C produces also a significant CO2 uptake, even larger than that previously observed for the CeO2 control. Table 1 provides the amount of CO2 adsorbed on Au/CeO2. However, in the case of Au/CeO2 dosed with hydrogen, incorporation of CO2 produced significant spectroscopic changes as shown in Fig. 3 and 4. As can be seen in these figures, the relatively narrow peak from 400 to 600 cm−1 disappears and is replaced by a broad band from 500 to 650 cm−1 peaking at 550 cm−1. These spectral variations indicate the reaction of the bridged hydroxyl groups generated by hydrogen chemisorption on the Au/CeO2 surface with CO2 at 150 °C, a fact that agrees with the reported catalytic activity.39–41Eqn (3) proposes a reaction compatible with the reported information and with the INS as well as the IR spectral changes discussed below.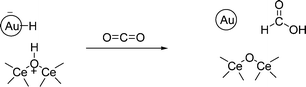 | (3) |
Besides CO2 hydrogenation, gold catalysts have also been used for the formation of H2O2 by reaction of hydrogen with oxygen.42–45 This is a process with potentially huge industrial impact for the in situ generation of H2O2 and its in-house use in other processes such as the formation of epoxides.46–49 Since our in situ spectroscopic study by INS has shown the appearance of new hydroxyl groups, it is of interest to determine the reactivity of these bridging hydroxyls on the catalyst surface with oxygen. Therefore, we also exposed the hydrogen dosed Au/CeO2 catalyst to O2. As may be seen from Table 1, the hydrogen-treated Au/CeO2 adsorbed a significant amount of oxygen at room temperature. Notably, neither hydrogen-treated CeO2 nor Au/CeO2 that has not been exposed to hydrogen adsorb a significant amount of oxygen. Thus, volumetric data suggest that the O2 uptake is due to the reaction of O2 with the hydrogen species generated in the chemisorption of hydrogen. This was confirmed by INS spectroscopy.
Exposure of the hydrogen-treated Au/CeO2 to O2 at ambient temperature was sufficient to change the INS spectrum of the sample completely. It is interesting to note that according to the volumetric data, the oxygen uptake in the Au/CeO2 corresponds to half the moles of hydrogen, while oxygen adsorption in CeO2 is negligible. Fig. 5 shows the difference INS spectra indicating the reaction of the hydrogen-treated Au/CeO2 sample with O2. A very weak band is observed at 180 cm−1, which is provisionally assigned to the torsion about the Au–O bond of an adsorbed hydroperoxide, OOH. This mode is visible in the INS spectrum, despite being on the gold, because the torsion results in a large amplitude motion of the hydrogen and hence significant INS intensity. The bands attributed to hydroperoxide formation as reported by Goodman et al. on Au/TiO2 were not detected.32 In contrast to the present case, we note that there was a large amount of water present in the reported INS spectrum of Au/TiO232 and the band at 1230 cm−1 may be due to a H2O2–H2O complex rather than a surface species.
IR spectroscopy study
INS is a very appropriate technique to monitor hydrogen species in or on solids.50 However, due to the small cross-section of other nuclei to neutrons, INS spectroscopy is less informative with respect to other functional groups. In particular, the small percentage of Au in the sample results in a lack of sensitivity for the detection of gold hydrides. Also the fate of adsorbed CO2 reacting with bridged hydroxyls and the possible formation of carbonates on the catalyst surface can go undetected by INS spectroscopy. For this reason, and in order to gain additional information on the chemisorption processes, we performed a parallel in situ spectroscopic study complementing the INS study with IR spectroscopy.The results of hydrogen chemisorption on Au/CeO2 are presented in Fig. 6. Upon outgassing Au/CeO2 for 2 h at 150 °C, the IR spectrum reveals the presence of two hydroxyl bands, one sharper at 3656 cm−1 overlapping with a broader band from about 3600 cm−1 to 3300 cm−1. The IR spectrum also shows vibrations in the 1600–1400 cm−1 region, attributable, at least in part, to the presence of carbonates on the catalyst.
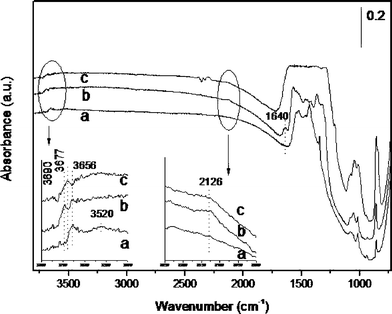 | ||
| Fig. 6 IR spectra recorded at 23 °C for the Au/CeO2 sample after outgassing at 150 °C for 2 h (a), subsequent exposure to H2 at 150 °C for 2 h (b); and exposing the hydrogen-dosed Au/CeO2 sample to CO2 at 150 °C for 2 h (c). Inset: expanded IR spectra. | ||
When the outgassed Au/CeO2 sample was contacted with hydrogen at 150 °C for 2 h, the resulting IR spectrum showed changes in the hydroxyl stretching vibration compatible with the INS study previously discussed. Thus, chemisorption of hydrogen leads to the appearance of a new relatively narrow hydroxyl band at 3690 cm−1 while the peak at 3656 cm−1 is maintained. Formation of water can also be deduced from the appearance of the 1640 cm−1 band and is compatible with the previous comments on the hydrogen chemisorption measurements. But, the most important observation was the appearance of a new peak at 2134 cm−1 that is assigned to the Au–H stretching mode of a gold hydride. Previous reports have observed the Au–H peak at 2164 cm−1, which is close to the present wavenumber.22 These observations are compatible with eqn (1) and (2). Control experiments involving IR spectroscopy on CeO2 failed to lead to detectable spectral changes, in agreement with TPR, Table 1 and the INS study.
In order to provide additional evidence in support of the assignment of the 2134 cm−1 band to Au–H, experiments using D2 instead of H2 were carried out. Under these conditions, we observed a considerable decrease in the intensity of the residual Au–H band. According to the atomic weights, the expected position of the Au–D at 1515 cm−1 (νAu–H/1.4107) falls on top of the strong ceria framework vibrations and is undetectable. Also the O–H vibration bands at 3690 and 3656 cm−1 disappear or decrease to a significant extent and the appearance of the corresponding O–D vibrations at 2698 and 2666 cm−1 (νO–H/1.3745) is observed. Expansions of the relevant region of the IR spectra recorded using H2 or D2 as reagent are provided in the ESI (Fig. S1†). The failure to observe a complete disappearance of the hydrogen bands in the isotopic labeling experiments is due to the large concentration of 1H atoms in the starting sample.
When this hydrogen-treated Au/CeO2 catalyst was exposed to CO2 at 150 °C for 2 h a decrease in the intensity and peak area of the gold hydride band occurred, accompanied by changes in the hydroxyl region. Particularly, the narrow vibrational band at 3619–3656 cm−1 disappeared or shifted towards lower frequency. Simultaneously the 1600–1400 cm−1 region became saturated indicating the massive formation of carbonates. Unfortunately the formation of reduced products from CO2 such as formate could not observed due to the overlap of the most intense carbonyl stretching vibrations with the carbonate bands. Thus, the data presented in Fig. 6 provide evidence for the reaction of Au–H with CO2 as indicated in eqn (3), accompanied by the massive formation of carbonate masking the bands of formate. Interestingly, carbonate formation also occurs on CeO2 and many other solids (particularly basic solids) and the formation of metal carbonates on the particle surface is well documented.38,51,52 Thus, two independent and parallel processes, reaction of Au–H and carbonate formation on the support, take place when hydrogen-treated Au/CeO2 is exposed to CO2.
The reactivity towards O2 of the hydrogen-treated Au/CeO2 sample was also studied by FTIR spectroscopy. Prior FTIR studies with Au catalyst have already observed the 830 cm−1 band and attributed it to ionic HO2− peroxide interacting with positive gold.53,54 IR spectra of the oxygen-dosed, hydrogen-treated Au/CeO2 also show changes in the hydroxyl region, but as in the case of the CO2 absorption, the information from IR in this region is less clear than that obtained by INS. The results are shown in Fig. 7.
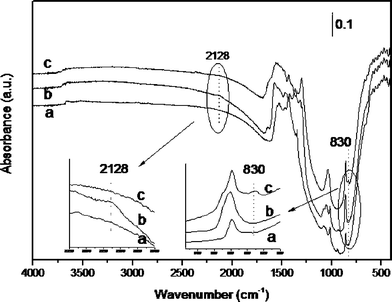 | ||
| Fig. 7 IR spectra recorded at 23 °C for the Au/CeO2 sample after outgassing at 150 °C for 2 h (a), subsequent exposure to H2 at 150 °C for 2 h (b); and exposing the hydrogen-dosed Au/CeO2 sample to O2 at 23 °C for 1 h (c). Inset: expanded zones of the IR spectra. | ||
As in the previously discussed case, we started by incorporating hydrogen onto carefully dehydrated Au/CeO2. Subsequently the hydrogen-dosed Au/CeO2 sample was exposed to an oxygen atmosphere. As Fig. 7 shows, the Au–H peak at 2128 cm−1 that is generated upon hydrogen chemisorption at 150 °C on Au/CeO2, disappears completely when oxygen is admitted into the cell at room temperature and atmospheric pressure. Concomitantly with the disappearance of the Au–H, the formation of a peak at 830 cm−1 was observed. According to reports in the literature, this 830 cm−1 peak can be assigned to the O–OH vibration,53,54 and is present in the IR spectrum of hydrogen peroxide in the gas phase.32 This assignment is compatible with the observation by INS spectroscopy of a weak 180 cm−1 band attributable to the torsion of the Au–O in adsorbed hydroperoxide.
The assignment of the O–OH vibration was also supported by performing an alternative experiment using isotopically enriched 18O2. In agreement with the assignment, when 18O2 was used as reagent the 830 cm−1 peak was no longer observed and was replaced by the corresponding 18O–18OH vibration at the predicted 782 cm−1 (ν16O–16O/1.0606) wavenumber. Expansions of the relevant region of the IR spectra recorded using O2 and 18O2 are provided in the ESI (Fig. S2†).
A reasonable proposal to rationalize the spectroscopic observations upon reaction with O2 is illustrated in eqn (1) and (2). Interestingly, neither our neutron scattering nor IR spectra have allowed the detection of the 1230 cm−1 band that has been previously observed by INS spectroscopy on the surface of a Au/TiO2 sample acting as the catalyst for hydrogen peroxide formation from H2 and O2 at 150 °C after rapid quenching by liquid nitrogen.32
Conclusions
The present study provides data showing again the unique features of gold nanoparticles supported on nanoparticulate ceria. INS and IR spectroscopy provide data showing that the remarkable hydrogen chemisorption on Au/CeO2 results in a heterolytic bond cleavage leading to the concomitant formation of Au–H and positively charged bridged OH groups, besides the generation of water from framework oxygens. Thus, there is a dissymmetric location of the two hydrogen atoms in the H2 splitting, one bonded to the ceria support oxygen and the other on the gold nanoparticles. The current paradigm in noble metal hydrogenation considers that hydrogen is dissociated on the metal nanoparticles and remains there as metal hydride (homolytic cleavage) or in a later stage can spill over the solid surface. In the present case, we have shown conclusively by TPR that the support and gold are necessary to observe low-temperature hydrogen chemisorption and that the species formed contain one hydrogen atom bonded to gold, but the other is bonded to oxygen atoms of the support. The assignments have been supported by the spectroscopic variations observed in the measurements using isotopes. Our work is of relevance for heterogeneous catalysis by gold and can serve to rationalize current data and develop new processes.Experimental section
Samples
Au/TiO2 was a commercial sample supplied by the World Gold Council (reference A catalyst). CeO2 was prepared as reported previously by hydrolysis under controlled pH of Ce(NO3)3.30 Au/CeO2 was obtained by the deposition–precipitation method by suspending CeO2 in aqueous HAuCl4 solutions at controlled pH.33 CeO2 and Au/CeO2 are exactly the same batches as those used for the CO2 fixation by ω-aminoalcohols.38Inelastic neutron scattering spectroscopy
INS spectroscopy is a complementary form of vibrational spectroscopy, where the scattering event is between a neutron and the atomic nucleus, thus the electronic nature of the material, conductor, semiconductor or insulator, is irrelevant. The scattering intensity depends on the incoherent inelastic scattering cross-section and the amplitude of vibration. For 1H both of these are large, consequently the scattered intensity is dominated by hydrogenous motion. The INS experiments were performed with the high resolution time-of-flight spectrometer TOSCA at the ISIS pulsed spallation neutron source at the STFC Rutherford Appleton Laboratory, Chilton, UK. The instrument has been described in detail previously.55 In brief TOSCA is a pulsed neutron, indirect geometry, low band-pass broadband (25–4000 cm−1) spectrometer with good energy resolution (ΔEt/Et ≈ 1.25%). All the INS measurements were made at 20 K using a closed cycle refrigerator to cool the sample, the measurement time was typically 12 h per spectrum. The catalyst (20 g nanoparticulate CeO2 or 19 g Au/CeO2) was loaded into an Inconel sample can. The can was attached to a gas manifold and the sample was evacuated at 150 °C for one hour. The INS spectrum was then recorded and used as the background for subsequent measurements. The sample was then reacted with H2 gas at 150 °C for one hour and then with CO2 at 150 °C for one hour, the INS spectrum was recorded after each treatment. This procedure was then repeated except that O2 was used in the final step rather than CO2. At each stage the gas uptake was recorded and these are reported in Table 1 after correction for the dead volume in the system.FT IR spectra
FTIR spectra were collected on a BioRad FTS-40A spectrometer. The infrared cell, connected to a dosing system, was designed to treat the samples in situ under controlled atmospheres and temperatures. The samples were evacuated at 10−5 mbar and 373 K for 1 h prior to the adsorption experiments. Chemisorption of H2, CO2 and O2 was performed following the same protocol as previously discussed for the INS measurements. After reactant adsorption the sample was evacuated in order to remove the excess of both reactants. Isotopic studies were carried out under exactly the same conditions using D2 and 18O2.Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation (CTQ2009-11583) is gratefully acknowledged. The STFC Rutherford Appleton Laboratory is thanked for access to neutron beam facilities.Notes and references
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- S. A. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef.
- G. C. Bond, Catal. Today, 2002, 72, 5–9 CrossRef CAS.
- G. C. Bond, C. Louis and D. T. Thompson, Catalysis by Gold, Imperial College Press, London, 2007 Search PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- M. Haruta, Gold Bull. (London, U. K.), 2004, 37, 27–36 Search PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS.
- N. R. Shiju and V. V. Guliants, Appl. Catal., A, 2009, 356, 1–17 CrossRef CAS.
- M. Boronat, P. Concepcion, A. Corma, S. Gonzalez, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230–16237 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS.
- A. Corma, P. Serna and H. Garcia, J. Am. Chem. Soc., 2007, 129, 6358–6359 CrossRef CAS.
- G. C. Bond, P. A. Sermon, G. Webb, D. A. Buchanan and P. B. Wells, J. Chem. Soc., Chem. Commun., 1973, 444b–445 RSC.
- G. C. Bond and D. T. Thompson, Catal. Rev. Sci. Eng., 1999, 41, 319–388 CrossRef CAS.
- A. Abad, C. Almela, A. Corma and H. Garcia, Chem. Commun., 2006, 3178–3180 RSC.
- P. Serna, M. Lopez-Haro, J. J. Calvino and A. Corma, J. Catal., 2009, 263, 328–334 CrossRef CAS.
- L. Andrews, Chem. Soc. Rev., 2004, 33, 123–132 RSC.
- G. N. Khairallah, R. A. J. O'Hair and M. I. Bruce, Dalton Trans., 2006, 3699–3707 RSC.
- X. F. Wang and L. Andrews, Angew. Chem., Int. Ed., 2003, 42, 5201–5206 CrossRef CAS.
- A. Escalle, G. Mora, F. Gagosz, N. Mezailles, X. F. Le Goff, Y. Jean and P. Le Floch, Inorg. Chem., 2009, 48, 8415–8422 CrossRef CAS.
- H. Ito, T. Saito, T. Miyahara, C. M. Zhong and M. Sawamura, Organometallics, 2009, 28, 4829–4840 CAS.
- M. Boronat, F. Illas and A. Corma, J. Phys. Chem. A, 2009, 113, 3750–3757 CrossRef CAS.
- A. Corma, M. Boronat, S. Gonzalez and F. Illas, Chem. Commun., 2007, 3371–3373 RSC.
- A. Grirrane, A. Corma and H. Garcia, Science, 2008, 322, 1661–1664 CrossRef CAS.
- A. Abad, P. Concepcion, A. Corma and H. Garcia, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS.
- S. Carrettin, P. Concepcion, A. Corma, J. M. Lopez Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS.
- A. Abad, C. Almela, A. Corma and H. Garcia, Tetrahedron, 2006, 62, 6666–6672 CrossRef CAS.
- V. B. Kazansky, I. R. Subbotina, R. A. van Santen and E. J. M. Hensen, J. Catal., 2004, 227, 263–269 CrossRef CAS.
- C. Sivadinarayana, T. V. Choudhary, L. L. Daemen, J. Eckert and D. W. Goodman, J. Am. Chem. Soc., 2004, 126, 38–39 CrossRef CAS.
- A. Abad, A. Corma and H. Garcia, Chem.–Eur. J., 2008, 14, 212–222 CrossRef CAS.
- R. Juarez, A. Corma and H. Garcia, Green Chem., 2009, 11, 949–952 RSC.
- A. Grirrane, A. Corma and H. Garcia, J. Catal., 2009, 264, 138–144 CrossRef CAS.
- A. Corma, P. Atienzar, H. Garcia and J.-Y. Chane-Ching, Nat. Mater., 2004, 3, 394–397 CrossRef CAS.
- M. Aresta, A. Dibenedetto, C. Pastore, C. B. Corrado, A. B. Brunella, S. Cometa and E. De Giglio, Catal. Today, 2008, 137, 125–131 CrossRef CAS.
- R. Juárez, P. Concepción, A. Corma and H. García, Chem. Commun., 2010, 46, 4181–4183 RSC.
- R. A. Koeppel, A. Baiker, C. Schild and A. Wokaun, J. Chem. Soc., Faraday Trans., 1991, 87, 2821–2828 RSC.
- H. Sakurai and M. Haruta, Appl. Catal., A, 1995, 127, 93–105 CrossRef CAS.
- H. Sakurai, S. Tsubota and M. Haruta, Appl. Catal., A, 1993, 102, 125–136 CrossRef CAS.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2008, 138, 225–239 RSC.
- P. Landon, P. J. Collier, A. F. Carley, D. Chadwick, A. J. Papworth, A. Burrows, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1917–1923 RSC.
- J. K. Edwards, B. E. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058–2059 RSC.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- G. Mul, A. Zwijnenburg, B. van der Linden, M. Makkee and J. A. Moulijn, J. Catal., 2001, 201, 128–137 CrossRef CAS.
- T. A. Nijhuis, B. J. Huizinga, M. Makkee and J. A. Moulijn, Ind. Eng. Chem. Res., 1999, 38, 884–891 CrossRef CAS.
- E. E. Stangland, K. B. Stavens, R. P. Andres and W. N. Delgass, J. Catal., 2000, 191, 332–347 CrossRef CAS.
- P. C. H. Mitchell, S. F. Parker, A. J. Ramirez-Cuesta and J. Tomkinson, Vibrational spectroscopy with neutrons, with applications in chemistry, biology, materials science and catalysis, World Scientific, Singapore, 2005 Search PubMed.
- Y. Himeda, Eur. J. Inorg. Chem., 2007, 3927–3941 CrossRef CAS.
- L. Znak, K. Stolecki and J. Zielinski, Catal. Today, 2005, 101, 65–71 CrossRef CAS.
- A. A. Gewirth, X. Li and J. Kim, Abstracts of Papers of the American Chemical Society, 2006, 231, 616 –INOR.
- J. W. Kim and A. A. Gewirth, J. Phys. Chem. B, 2006, 110, 2565–2571 CrossRef CAS.
- D. Colognesi, M. Celli, F. Cilloco, R. J. Newport, S. F. Parker, V. Rossi-Albertini, F. Sacchetti, J. Tomkinson and M. Zoppi, Appl. Phys. A, 2002, 74 [Suppl.], S64–S66 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra of Au/CeO2 catalysts and samples. See DOI: 10.1039/c0sc00336k |
| This journal is © The Royal Society of Chemistry 2010 |