Highly selective formation of unsaturated esters or cascade reactions to α,ω-diesters by the methoxycarbonylation of alkynes catalysed by palladium complexes of 1,2-bis(ditertbutylphosphinomethyl)benzene†
A. Alberto
Núñez Magro
a,
Lynzi-Marie
Robb
a,
Peter J.
Pogorzelec
a,
Alexandra M. Z.
Slawin
a,
Graham R.
Eastham
b and
David J.
Cole-Hamilton
*a
aEaStCHEM, School of Chemistry, University of St. Andrews, St. Andrews, Fife, KY16 9ST, Scotland, UK. E-mail: djc@st-and.ac.uk; Fax: +44 (0)1334 463808; Tel: +44(0)1334 463805
bLucite International, Technology Centre, PO Box 90, Wilton, Middlesbrough, Cleveland, England, UK TS6 8JE. E-mail: graham.eastham@lucite.com.; Fax: +44 (0)1642 447109; Tel: +44 (0)1642 447109
First published on 14th October 2010
Abstract
The methoxycarbonylation of phenylethyne catalysed by Pd/1,2-bis-(ditertiarybutylphosphinomethyl)benzene gives the unusual linear product, methyl cinnamate with high activity (initial turnover frequency, TOFo > 1700 mol product·(mol catalyst·h)−1) and regioselectivity (>90%). Terminal aliphatic alkynes give α,β–unsaturated esters after short reaction times or α,ω-diesters, including dimethyl 1,6-hexanedioate (dimethyl adipate), from 1-butyne after longer times. The diesters are formed by a cascade methoxycarbonylation–isomerisation–methoxycarbonylation sequence. Methoxycarbonylation of internal alkynes (e.g. 4-octyne) leads to the formation of the mono-carbonylated product as a result of the low propensity of the tri-substituted double bond of the product towards isomerisation. Hydroxycarbonylation of phenylethyne gives predominantly E-3-phenylpropanoic acid with smaller amounts of branched and disubstituted products as well as 3-phenylpropanoic acid. Evidence is presented that the reactions occur via a hydride mechanism.
Introduction
Alkoxycarbonylation, which can be used for the synthesis of esters from a substrate (such as alkene, alkyne or halo-arene), carbon monoxide and an alcohol, is of potential interest to industry as a result of the low cost and availability of the required feed stocks as well as the low generation of waste.1For alkynes, previous studies have highlighted high activities and a marked tendency towards branched regioselectivity (Fig. 1). The synthesis of methyl 2-methylpropenoate (methyl methacrylate) (1, Fig. 1, R = R′ = Me, branched) from propyne in a single step by Drent2 and others3, as well as the preparation of methyl 2-phenylpropenoate (methyl atropate) (2, Ar = Ph)2d,4 which is related to the preparation of the anti-inflammatory drug, Ibuprofen (via asymmetric hydrogenation of the unsaturated ester), are examples of industrially interesting applications of this process. Despite the attractive nature of the branched products, β-substituted methyl acrylates or methyl cinnamates (in the case of R = Ar) are also highly desirable. However, only a few catalysts show a preference towards the linear regioisomeric products.2d, 4j As a result of this, 3-arylpropenoate esters are prepared via the Heck reaction,5a–c which, despite being a diverse and useful process, requires halo-arene starting materials and at least one equivalent of base, which has to be removed as waste from the reaction.
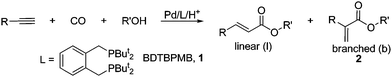 | ||
| Fig. 1 General scheme for the alkoxycarbonylation of alkynes. | ||
In this paper we report very high selectivities to linear products in alkyne methoxycarbonylation reactions (Fig. 2). Phenylethyne gives the Heck products with high E selectivity, whilst aliphatic alkynes first give α,β-unsaturated esters but then α,ω–diesters via an unprecedented methoxycarbonylation–isomerisation–methoxycarbonylation cascade sequence in which the second carbonylation only occurs when the double bond is in the thermodynamically least favoured terminal position
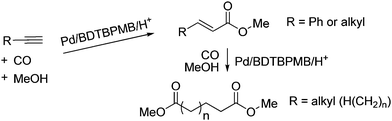 | ||
| Fig. 2 The linear carbonylation of alkynes to α,β–unsaturated esters, including Heck products, or α,ω-diesters, reported in this paper. | ||
Results and discussion
A process for the methoxycarbonylation of ethene to methylpropanoate has recently been commercialised as part of a new route to methylmethacrylate.6 It uses palladium complexes of 1,2-bis(ditertbutylphosphinomethyl)benzene (BDTBPMB) (1), for which we now report the X-ray structure (see ESI†). We have explored the chemistry of this system and find it to show very high activity and linear selectivity even under very mild conditions (room temperature, 1 bar) in the methoxycarbonylation of terminal or internal alkenes7,8 or of unsaturated esters, even if the double bond is conjugated to the ester or buried deep in the chain.8a Butadiene also gives the α,ω–diester, dimethyl 1,6-hexanedioate.8i Unusually, vinyl acetate,9 and styrene (89%)9b give high branched selectivity.Because of the usually very high linear selectivity obtained with Pd/BDTPBMB, we tested it in the methoxycarbonylation of alkynes. Since the linear products of methoxycarbonylation of linear terminal alkynes should be α,β–unsaturated esters, they should be further carbonylated to α,ω–diesters.8a
Methoxycarbonylation of phenylethyne
The methoxycarbonylation of phenylethyne (Fig. 3), is very fast and much faster than that of styrene. Indeed, for phenylethyne, the reaction is initially zero order in substrate, perhaps suggesting mass transport limitations in getting the gas into the system fast enough. Phenylethyne (96%) also gives a much higher selectivity than styrene (69% under these conditions) towards the linear product, (Table 1 and Fig. 4).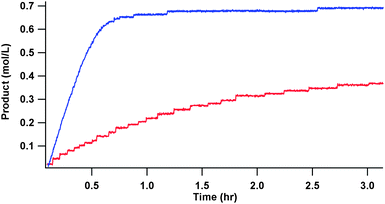 | ||
| Fig. 3 Product formation from methoxycarbonylation of phenylethyne (blue) and styrene (red) under the conditions of Table 1. The amount of product formed was determined from the pressure drop in a ballast vessel, from which CO was fed to maintain the pressure in the autoclave, assuming that the reaction only incorporates 1 mol of CO/mol of product generated. The reaction of styrene is not shown to completion owing to the length of time required. It did proceed to completion over 6.5 h. | ||
| Entry | Substrate | Rate Constant/s−1 | TOF0 (mol product·(mol catalyst·h)−1b | Linear Sel. (%)a |
|---|---|---|---|---|
a Conditions: Substrate (9.1 mmol), [Pd2dba3] (4.2 mg, 0.0046 mmol), BDTBPMB (22 mg, 0.055 mmol), MsOH/Pd ratio (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), MeOH (10 cm3), T = 80 °C, pCO = 30 bar, 3 h.
b measured by GC at the end of the reaction.
c In the first order region (0.54–0.64 h). :1), MeOH (10 cm3), T = 80 °C, pCO = 30 bar, 3 h.
b measured by GC at the end of the reaction.
c In the first order region (0.54–0.64 h).
|
||||
| 1 | Phenylethyne | 0.0009c | 1780 | 96 |
| 2 | Styrene | 0.0002 | 276 | 69 |
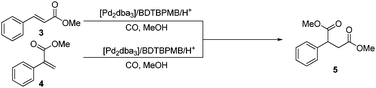 | ||
| Fig. 4 Possible origins of dimethyl 3-phenylbutanedioate, 5, during the methoxycarbonylation of phenylethyne. 5 was not observed when starting from 3. | ||
Further studies in a closed batch reactor (Table 2) also showed a high catalyst activity (>99.9% conversion in 3 h) and a marked preference towards the linear isomer, methyl cinnamate (3) (yield = 93%, linear selectivity = 99%), (Table 2, entry 1). Interestingly, α,β double carbonylation, forming dimethyl 2-phenyl-1,4-butandioate (5) (7%), also occurred to a small extent. No other dicarbonylation products were detected by GC. There was a decrease in the yield of 5 when the reaction was run at room temperature, the yield of branched product, 4, increased, and there was a decrease in the linear selectivity for the monoesters (Table 2, entry 2). Decreasing the carbon monoxide pressure resulted in a lower regioselectivity as well as a significant drop in yield, (Table 2, entry 3).
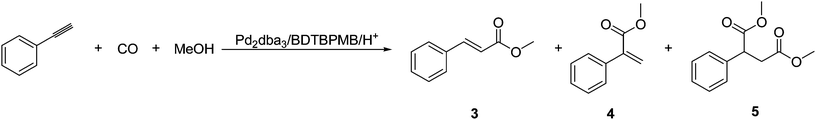
| Entry | [Pd2dba3] (mol %) | 1 (mol %) | T/°C | P/bar | T/h | Ester (3 + 4) yield (%) | Linear Sel. (%) | Diester 5 yield (%) | α,β Sel. (%) |
|---|---|---|---|---|---|---|---|---|---|
|
a Conditions: Phenylethyne (1 cm3, 9.1 mmol), [Pd2dba3] (as described), BDTBPMB (as described), MsOH/Pd ratio (30:1), MeOH (10 cm3).
b [BDTBPMBH2][BF4]2 used in place of BDTBPMB.
c No MsOH added.
|
|||||||||
| 1 | 0.25 | 3 | 80 | 30 | 3 | 93 | 99 | 7 | >99 |
| 2 | 0.25 | 3 | rt | 30 | 3 | 98 | 98 | 2 | >99 |
| 3 | 0.25 | 3 | 80 | 2 | 3 | 63 | 90 | 0 | — |
| 4 | 0.05 | 0.6 | 80 | 30 | 3 | >99 | 91 | 0 | — |
| 5 | 0.25 | 3 | 80 | 30 | 14 | 95 | 99 | 5 | >99 |
| 6 | 0.1 | 0.55b | 80 | 30 | 3 | 99 | 95 | ||
| 7 | 0.1 | 0.55b,c | 80 | 30 | 3 | 67 | 96 |
At lower palladium concentrations, the methoxy carbonylation of phenylethene showed higher conversion to 3; however, the dicarbonylation product, 5, was not obtained (Table 2, entry 4). The yield of 5 was also not increased by carrying out the reaction using a higher palladium loading over a longer time (14 h) (Table 2, entry 5). This may suggest that 5 is formed only from 4 and not from 3 so that, when all 4 has been carbonylated, no more 5 is formed
Methoxycarbonylations of 3 and 4 were carried out separately under the conditions used for phenylethyne. 4 gave 66% conversion to 5 (Table 3, entry 1), whilst 5 was not obtained from carbonylation of 3 (Table 3, entry 2). Even at higher catalyst loadings, only traces of 5 were formed from 3 (Table 3, entry 3). This therefore confirms that the formation of 5 is as a result of the methoxycarbonylation of 4, but not of 3 (Fig. 4).
BDTBPMB is air sensitive and is usually weighed in a glove box. In order to determine if the air stable protonated analogue of BDTBPMB has the capacity to generate the ligand for this reaction, the phosphonium salt, [BDTBPMBH2][BF4] (X-ray structure in ESI†) was synthesised, from BDTBPMB and HBF4 in diethylether.10
When the phosphonium salt was used in phenylethyne carbonylation, the yield and selectivity were similar to those obtained when using BDTBPMB. (Table 2, entry 6). Although the additional presence of a strong acid resulted in more efficient hydride formation (Table 2, entry 6), [BDTBPMBH2][BF4]/[Pd2dba3] worked successfully without the addition of excess acid (Table 2, entry 7), suggesting again9c that the Pd–H bond required can possibly be generated by oxidative addition of a P–H bond across Pd0. Such a step has been proposed before as being mechanistically important.6g
The effects of alternative ligands, metal precursors and alcohol nucleophiles on the methoxycarbonylation of phenylethyne are included in the ESI.†
Methoxycarbonylation of linear aliphatic alkynes
We have previously shown that the isomerisation of the terminal double bond along a long chain is fast when the reaction is catalysed by [Pd2dba3]/BDTBPMB/H+ under CO.7,8a This can be used for the tandem generation of α,ω-diesters by alkoxycarbonylation of unsaturated esters even if the double bond is conjugated with the ester group.8a, b We, therefore, reasoned that it should be possible to form an α,ω-diester in a single cascade reaction via the methoxycarbonylation of linear aliphatic alkynes. It was expected that the first carbonylation would generate an α,β-unsaturated ester, and its double bond could then isomerise to the end of the chain, before undergoing a second carbonylation in the terminal position. High regioselectivity was expected since it has been shown that the methoxycarbonylation of unsaturated esters gives α,ω–diesters regardless of the initial position of the double bond.8a, cUnder the conditions used for the methoxycarbonylation of phenylethyne (0.1 mol % Pd, 30 bar, 80 C), 1-butyne reacts extremely rapidly (gas transport limited) to give methyl 2-pentenoate, together with unconjugated linear isomers and small amounts of the branched product, methyl 2-methyl-2-butenoate (Table 4, entry 1), but after longer reaction times, dimethyl 1,6-hexandioate (dimethyladipate, 8, m = 1)), one of the precursors to nylon 6.6, is obtained with excellent yield and selectivity (Table 4, entry 2). 1-Pentyne reacted more slowly to give predominantly methyl 2-hexenoate (6, m = 2) after 3 h (Table 4, entry 3), although significant amounts of esters with the double bond in other positions in the chain (7, n + p = 2, p ≠ 0) were also observed. Dimethyl 1,7-heptanedioate (8, m = 2) was not obtained under these conditions. The generation of dimethyl 1,7-heptanedioate was, however, achieved in excellent yield (>99%) and regioselectivity (92%) by increasing the concentration of the palladium complex in the medium to 0.5 mol %. (Table 4, entry 4).
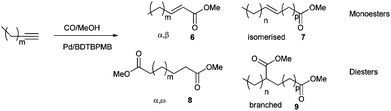
| Entry | Substrate | [Pd2dba3] (mol %) | T/h | Ester yield (6 + 7) (%) | α,β-Unsaturated ester (6) (%) | Diester yield (8 + 9) (%) | α,ω–Diester (8) (%) |
|---|---|---|---|---|---|---|---|
|
a Conditions: Substrate (9.1 mmol), [Pd2dba3] (as indicated), BDTBPMB/Pd mol ratio (6:1), MsOH/Pd ratio (30:1), T = 80 °C, pCO = 30 bar, MeOH (10 mL).
|
|||||||
| 1 | 1-Butyne | 0.1 | 0.5 | 100 | 83 | ||
| 2 | 1-Butyne | 0.1 | 3 | >99 | >99 | ||
| 3 | 1-Pentyne | 0.05 | 3 | 90 | 73 | 0 | 0 |
| 4 | 1-Pentyne | 0.25 | 3 | 0 | 0 | >99 | 92 |
| 5 | 1-Octyne | 0.05 | 3 | 87 | 75 | 0 | 0 |
| 6 | 1-Octyne | 0.25 | 3 | 34 | 29 | 66 | 61 |
| 7 | 1-Octyne | 0.5 | 3 | 0 | 0 | >99 | 85 |
| 8 | 1-Octyne | 0.25 | 14 | 0 | 0 | >99 | 87 |
Using 1-octyne, under the same conditions of lower Pd loading, no diester was formed after 3 h but only methyl 2-nonenoate (6, m = 5) together with small amounts of other isomers with the double bond isomerised along the chain (7, n + p = 5, p ≠ 0), Table 4, entry 5). Increasing the palladium concentration to 0.5 mol % yielded some dimethyl 1,10-decanedioate (8, m = 5) with good regioselectivity (Table 4, entry 6) but full conversion to the α,ω–diester was only obtained with a Pd loading of 0.5 mol % or by extending the reaction time to 14 h. In these cases, the selectivity to the α,ω–diester was of the order of 85% (Table 4, entries 7 and 8). The other products were diesters with at least one ester group on an internal C atom (9, n + p = 5).
The kinetics of the carbonylation of 1-pentyne and 1-octyne were measured at constant CO pressure and are presented in Fig. 5 as plots of CO consumed per mole of alkyne used. It is clear that the initial carbonylation of the alkene is extremely fast. For 1-octyne, this reaction is complete in 1.5 mins and is probably mass transport limited. At this point, the products are unsaturated monoesters with the methyl hex-2-enoate (62%) dominating, but the other isomers with double bonds isomerised along the chain also being observed. There is also a small amount of methyl 2-methylpent-2enoate (10%).
![Kinetic plots at constant CO pressure for the carbonylation of 1-pentyne and 1-octyne. Inset are the same plots over the first 20 mins. Conditions: [Pd2(dba)3] (21mg, 0.023 mmol), BDTBPMB (110mg, 0.28 mmol), alkyne (1 cm3, 1-pentyne 0.01 mol, 1-octyne 0.0072 mol), MeSO3H (45 μl, 0.46 mmol), methanol (10 cm3), 80 °C, pCO = 30 bar.](/image/article/2010/SC/c0sc00276c/c0sc00276c-f5.gif) | ||
| Fig. 5 Kinetic plots at constant CO pressure for the carbonylation of 1-pentyne and 1-octyne. Inset are the same plots over the first 20 mins. Conditions: [Pd2(dba)3] (21mg, 0.023 mmol), BDTBPMB (110mg, 0.28 mmol), alkyne (1 cm3, 1-pentyne 0.01 mol, 1-octyne 0.0072 mol), MeSO3H (45 μl, 0.46 mmol), methanol (10 cm3), 80 °C, pCO = 30 bar. | ||
The second carbonylation is very much slower, hence accounting for the ability to have high selectivity to unsaturated monoesters after short reaction times, but proceeds to completion over about 20 h, when the reaction product is predominantly the α,ω–diester (for 1-octyne), 84% with 12% branched ester and 4% unsaturated ester. In both cases the fast gas uptake stops after about 0.9 mol of CO has been consumed. The plots assume that all the substrate enters the reactor when it is injected from the injector, but separate studies have shown that small amounts are held up in the injector. The final levelling off of the gas uptake at around 1.8 and 1.7 moles of CO for 1-octyne and 1-pentyne respectively would also be consistent with this explanation. In addition, the reactions give exotherms, warming the solutions by up to 12 °C and are so fast that some initial gas uptake may not be measured in the time taken for the reaction to stabilise.
Methoxycarbonylation of other alkynes
The methoxycarbonylation of 2-butyne, an internal alkyne gave a mixture of the two dicarbonylation products, dimethyl 2-methylpentane-1,5-dioate and dimethyl 2-ethylbutane-1,4-dioate. If the reaction is stopped after 3 h small amounts of methyl 2-methylbutenedioates are observed (Fig. 6). Evidently, the double bond in the intermediate unsaturated ester migrates to either end of the chain where it is carbonylated. Isomerisation away from the quaternary centre rather than past it is preferred. 4-Octyne, on the other hand, yielded exclusively the α,β-unsaturated mono ester, methyl 2-propylhex-2-enoate, (Fig. 6). No isomers of this product were detected, presumably because the tri-substituted conjugated double bond is thermodynamically favoured. The failure to observe dicarbonylation products suggests that the equilibrium concentration of terminal alkene as a result of isomerisation is vanishingly small.![Methoxycarbonylation of a) 2-butyne (3 or 20 h) and b) 4-octyne (4 h) catalysed by [Pd2dba3]/BDTBPMB/H + [Pd2dba3] (0.05 mol %), BDTBPMB (0.6 mol %), MsOH (3 mol %), pCO = 30 bar, MeOH (10 ml), 80 °C.](/image/article/2010/SC/c0sc00276c/c0sc00276c-f6.gif) | ||
| Fig. 6 Methoxycarbonylation of a) 2-butyne (3 or 20 h) and b) 4-octyne (4 h) catalysed by [Pd2dba3]/BDTBPMB/H + [Pd2dba3] (0.05 mol %), BDTBPMB (0.6 mol %), MsOH (3 mol %), pCO = 30 bar, MeOH (10 ml), 80 °C. | ||
1-Phenyl-1-propyne gave high conversion to monoesters but with low regioselectivity (Table 5, entry 1), as expected for the internal triple bond, but more surprisingly the methoxycarbonylation of the triple bond in 3-phenyl-1-propyne also gave low regioselectivity (Table 5, entry 2).

| Entry | R | R` | 10 yield (%) | 11 yield (%) |
|---|---|---|---|---|
|
a Conditions: Substrate (9.1 mmol), [Pd2dba3] (4.2 mg, 0.0046 mmol), BDTBPMB (22 mg, 0.055 mmol), MsOH/Pd ratio (30:1), T = 80 °C, pCO = 30 bar, MeOH (10 cm3), 3 h.
b Methyl 4-phenylbut-3-enoate (2%) was also obtained.
|
||||
| 1 | Ph | Me | 60 | 40 |
| 2 | Bz | H | 66 | 29b |
Trimethylsilylethyne gave dimethyl 1,4-butanedioate (12) and its regioisomer, dimethyl 2-methypropane-1,3-dioate (13) (Fig. 7), offering a potential alternative route to diesters from alkynes.
![Methoxycarbonylation of trimethylsilylethyne. [Pd2dba3] (0.05 mol %)/BDTBPMB (0.6 mol %), MsOH (3 mol %), pCO = (30bar), MeOH (10 cm3), 80 °C, 3 h.](/image/article/2010/SC/c0sc00276c/c0sc00276c-f7.gif) | ||
| Fig. 7 Methoxycarbonylation of trimethylsilylethyne. [Pd2dba3] (0.05 mol %)/BDTBPMB (0.6 mol %), MsOH (3 mol %), pCO = (30bar), MeOH (10 cm3), 80 °C, 3 h. | ||
Hydroxycarbonylation of phenylethyne
Hydroxycarbonylation reactions are usually significantly slower than methoxycarbonylation reactions and result in some catalyst deactivation.1 Our studies on palladium BDTBPMB for the hydroxycarbonylation of alkenes showed low rates and required high pressures of CO.11 In hydroxycarbonylation of phenylethyne catalysed by Pd/BDTBPMB, there was a reasonably high conversion at low palladium concentrations (although significantly lower than the conversions shown for methoxycarbonylation), with the major product being E-3-phenylpropenoic acid (14) (Table 6, entry 1). 2-Phenyl-1,4-butanedioic acid, 16, was also detected together with 3-phenylpropanoic acid, 17 (3%). This saturated product presumably arises from hydrogenation of the major product, 3-phenylpropenoic acid, 14. There is some precedent for this hydrogenation in the predominant formation of dimethyl 1,19-nonanediote in the methoxycarbonylation of methyl linoleate (2 double bonds) or methyl linolenate (3 double bonds) using the same catalytic system.8a For the reaction carried out in water, the hydrogen required for saturation to occur may be formed by a water gas shift reaction.1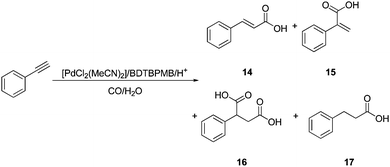
| Entry | Diphosphine | Acid yield (%) | Linear Sel. (%) | 16 (%) | 17 (%) |
|---|---|---|---|---|---|
|
a Conditions: Phenylethyne (1 cm3, 9.1 mmol), [Pd(MeCN)2Cl2] (23.6 mg, 0.091 mmol), diphosphine (0.46 mmol), MsOH/Pd ratio (30:1), T = 80 °C, pCO = 70 bar, dioxane (10 cm3), water (2 cm3), 5 h.
|
|||||
| 1 | BDTBPMB | 88 | 96 | 9 | 3 |
| 2 | dppb | 59 | 41 | 28 | 13 |
| 3 | BDPPMB | 13 | 42 | 76 | 11 |
| 4 | BDTBPMF | 42 | 86 | 0 | 0 |
| 5 | [BDTBPMBH2][BF4]2 | 62 | >99 | 38 | 0 |
The use of bis(diphenylphosphino)butane dppb instead of BDTBPMB led to an increase in the formation of 16, but resulted in lower regioselectivity in the formation of the unsaturated monoesters, (Table 6, entry 2). The generation of the hydrogenated product, 17, increased. A larger amount of 16 was obtained when BDPPMB was used as the ligand, such that the diester became the major product. (Table 6, entry 3). There was also an increase in production of 17 (11%) using this ligand. BDTBPMF promoted significantly less activity than BDTBPMB giving only moderate conversion to 14 (Table 6, entry 4). Interestingly this ligand did not give 16 or 17. Unexpectedly, the use of the air-stable BDTBPMB analogue, [BDTBPMBH2][BF4]2, did not produce similar results to BDTBPMB (Table 6, entry 5). There was a significantly higher yield of 16 and no formation of 17, but the linear selectivity observed for the monoester was higher than when using BDTBPMB.
Origin of the high linear selectivity
The methoxycarbonylation of alkynes usually displays a marked preference for branched regioselectivity but our studies show that Pd/BDTBPMB leads mainly to linear products with E stereochemistry at the double bond.Two mechanisms, with a carbomethoxy group (Fig. 8, Path A) or hydride (Fig. 8, Path B) as the chain carrier, have been proposed for methoxycarbonylation reactions.
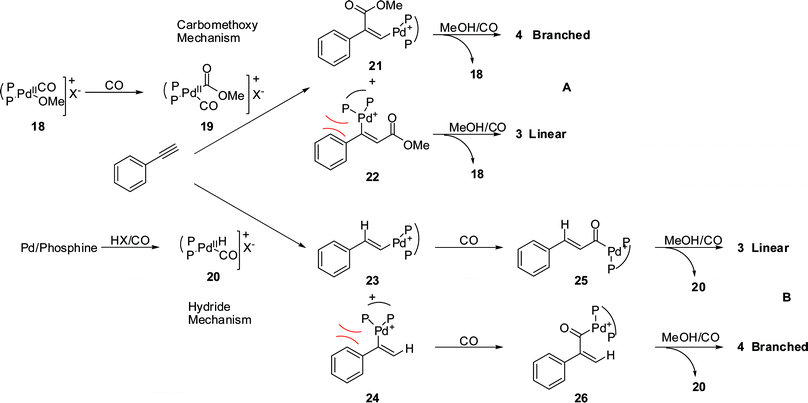 | ||
| Fig. 8 Carbomethoxy (A) and hydride (B) mechanisms for the methoxycarbonylation of phenylethyne. | ||
In the carbomethoxy mechanism (Fig. 8, Path A), the substrate reacts with the carbomethoxypalladium species 19 (formed via the reaction of the alkoxypalladium species 18 with CO) generating the intermediates 21 and 22. These react to produce the final linear and branched products by protonation with methanol.
In the hydride mechanism (Fig. 8, Path B), the first step involves the migration of the palladium hydride 20 onto the substrate forming species 23 and 24, which, via CO insertion and methanolysis, produce the final products 3 and 4. When the two mechanisms are compared, the C–C bond formation is significantly different. As a result of steric effects, the migration reactions would be expected to favour products in which the Pd atom and the phenyl group end up attached to different C atoms (intermediates 21 and 23). This predisposes the carbomethoxy mechanism to give the branched product, 4, via intermediate 21, whilst the hydride mechanism should favour the linear product, 3, via intermediate 23. In both cases cis addition (of Pd and CO2Me or of Pd and H) during the migratory insertion step should lead to the E isomer if the linear product, 3, is formed.
Methoxycarbonylation reactions catalysed by Pd/diphenyl-2-pyridylphosphine have been shown to follow the carbomethoxy mechanism with the pyridyl N atom acting as a proton shuttle and give branched products. 2a, b, 3a The high linearity observed when using Pd/BDTBPMB in the methoxycarbonylation of phenylethyne suggests that a hydride mechanism operates. The hydride mechanism has been proven to operate in the methoxycarbonylation of alkenes catalysed by the same system.6b, c, 7
Experimental
All experiments were carried out under dry argon using standard Schlenk line and catheter tubing techniques. Argon was dried through a Cr(II)/silica packed glass column. Liquids were transferred under inert atmosphere by syringe or canula though a septum. Solids were transferred directly from one Schlenk tube to another or weighed out in a glove box under argon.Carbon monoxide and hydrogen were purchased from BOC gases. Methanol was distilled over magnesium and stored under argon over molecular sieves. Acetonitrile, dioxane, ethanol and iso-propanol were degassed by the bubbling of argon and stored under argon over molecular sieves. Phenol was purchased from Sigma-Aldrich, recrystallised from petroleum (b. pt. 40–60 °C) and stored under argon. Water was distilled and degassed by the bubbling of argon, before storing under argon. Deuterated solvents were purchased from Sigma-Aldrich, degassed by the bubbling of argon, and stored under argon over molecular sieves.
[Pd(OAc)2]2 (Sigma-Aldrich) and [PdCl2] (Lancaster) were used as received. [Pt(MeCN)2Cl2] (Aldrich) and [Pd2dba3] (Lucite International) were stored under argon in a glove box. [PdCl2(MeCN)2]12 and [Pd(BDTBPMB)dba]6a were prepared according to the literature. Dppb (Sigma-Aldrich) and diphenyl-2-pyridylphosphine (Sigma-Aldrich) were stored under argon and used as received. BDPPMB was prepared according to literature.13 BDTBPMB and BDTBPMF were supplied by Lucite International and stored in a glove box. Methane sulfonic acid (Aldrich) was degassed under vacuum and stored under argon. Tetrabutylammonium fluoride and tetrabutylammonium iodide (Sigma-Aldrich) were stored in a desiccator and used as received. Phenylethyne, 1-phenyl-1-propyne, 3-phenyl-1-propyne, styrene, 1-butyne, 2-butyne, 1-pentyne, 1-octyne, 4-octyne and trimethylsilylethyne were purchased from Aldrich, degassed by the bubbling of argon (except in the case of the butynes) and stored under argon. Methyl E-3-phenylpropenoate (Aldrich) was used as received. Methyl 2-phenylpropenoate was prepared according to the literature.14 Tetrafluoroboric acid (Alfa Aesar) was used as received.
Batch autoclave reactions
[Pd2dba3] (4.2 mg, 0.0046 mmol) was placed in an autoclave, which was flushed three times with CO. BDTBPMB (22 mg, 0.055 mmol) was dissolved in methanol (10 cm3) in a degassed Schlenk flask. Alkyne (1 cm3, 9.1 mmol, or as stated in the Tables and Figure captions) and methane sulfonic acid were added to the solution, which was transferred to the autoclave via cannula. The autoclave was pressurised with CO (30 bar) and heated at 80 °C for 3 h. The autoclave was then cooled and vented. The solution was analysed by GCFID.Kinetic reactions
The apparatus included an autoclave, an injection arm, a pressure controller and transducer, a thermocouple pocket, a paddle stirrer, a ballast vessel, and a control panel. Each section could be isolated from the others with valves. The reaction took place in the autoclave, which was flushed with CO. [Pd2dba3] (4.2 mg, 0.0046 mmol) was placed in the autoclave, which was flushed three times with CO. BDTBPMB (22 mg, 0.055 mmol) was dissolved in methanol (10 cm3) in a degassed Schlenk flask. Methanesulfonic acid was added to the solution, which was transferred to the autoclave via cannula. Finally the autoclave was pressurised to 20 bar with CO. The stirrer was started and the autoclave was heated to 80 °C. Alkyne (1 cm3, 9.1 mmol, or as stated in the Tables and Figure captions) was added to the solution via the catalyst injector to initiate the reaction and the pressure adjusted to 30 bar. The gas consumed by the reaction was monitored by recording the drop in pressure in the ballast vessel, which fed gas to the reactor to retain the pressure at 30 bar. Data were collected via a link from data logging hardware (Pico Monitor, model ADC16) which was fitted to a PC though a COM port. The computer used data logging software (PicoLog for Windows, version 5.04.2) to monitor and record the pressures every 5 s. The data collected then allowed for the calculation of the kinetics of the reaction using the curve fitting programme, IgorPro. At the end of the reaction, the autoclave was cooled and vented. The solution was analysed by GCFID. For butynes, a cooled syringe was used to introduce the substrate into the catalyst injector.[BDTBPMBH2][BF4]2
BDTBPMB (2 g, 5 mmol) was dissolved in diethyl ether (20 cm3) in a degassed Schlenk flask. A solution of HBF4 (10 cm3, 50%–54% w/v in diethyl ether) was added dropwise. A white solid formed immediately. The solution was stirred for 3 h at room temperature. The solid was filtered in air, washed with diethyl ether (3 × 25 cm3) and acetone (3 × 25 cm3) and dried in vacuo (2.8 g, 96%). The white solid shows low solubility in all organic solvents with the exception of DMSO and DMF. It is stable in air in the solid state for several months but is quickly oxidised in solution.1H NMR (300 MHz, d8-DMSO, 298 K): 7.55 – 7.50 (m, 1 H), 7.43 – 7.37 (m, 1H), 3.97 (d, JPH = 13.8 Hz, 2 H), 1.37 (d, JPH = 16.3 Hz,18 H). 13C NMR (75 MHz, d8-DMSO, 298 K): 131.81, 131.73, 128.81, 33.47 (d, JPC = 35.8 Hz), 27.32, 18.70 (d, JPC = 38.0 Hz). 31P{1H}NMR (121 MHz, d8-DMSO, 298 K): 44.70 (bs). 19F{1H} NMR (282 MHz, CDCl3, 298 K): −143.91. m.p.: > 230 °C MS (EI+): Found: 395.2997. Required (M-H+): 395.3007. Despite repeated attempts, we were unable to obtain satisfactory microanalytical data for this compound, but its composition has been confirmed by an X-ray crystal structure.
Crystallographic data
X-ray diffraction studies for 1 and (1)PdCl2 were performed at 93 K using a Rigaku MM007/Mercury/diffractometer (confocal optics Mo-Kα radiation). Intensity data were collected using ω steps accumulating area detector frames spanning at least a hemisphere of reciprocal space for all structures (data were integrated using CrystalClear). All data were corrected for Lorentz, polarisation and long-term intensity fluctuations. Absorption effects were corrected on the basis of multiple equivalent reflections. Structures were solved by direct methods and refined by full-matrix least-squares against F2 (SHELXTL). All N–H and O–H hydrogen atoms were refined isotropically subject to a distance constraint (N,O–H = 0.98 Å). All remaining hydrogen atoms were assigned riding isotropic displacement parameters and constrained to idealised geometries.
1, colorless prisms, C24H44P2, M = 394.53, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.9447(19), b = 11.839(2), c = 11.991(2) Å α 117.625(6) β = 92.392(9) γ = 99.186(8)°, U = 1224.0(4) Å3, Z = 2, Dc = 1.070 Mgm−3, μ = 0.184 mm−1, F(000) = 436. Of 6891 measured data, 4039 were unique (Rint = 0.0199) and 3675 observed (I > 2σ(I)]) to give R1 = 0.0404 and wR2 = 0.1045, GOF = 1.040.
, a = 9.9447(19), b = 11.839(2), c = 11.991(2) Å α 117.625(6) β = 92.392(9) γ = 99.186(8)°, U = 1224.0(4) Å3, Z = 2, Dc = 1.070 Mgm−3, μ = 0.184 mm−1, F(000) = 436. Of 6891 measured data, 4039 were unique (Rint = 0.0199) and 3675 observed (I > 2σ(I)]) to give R1 = 0.0404 and wR2 = 0.1045, GOF = 1.040.
[BDTBPMBH2][BF4]2, C24H46P2(BF4)2, M = 528.94, Triclinic, space group P, a = 7.722(2), b = 15.101(4), c = 24.950(7) Å 90.779(7) β = 97.485(9) 92.145(10) °, U = 2882.0(13) Å3, Z = 4 (two independent molecules), Dc = 1.314 Mgm−3, μ = 0.215 mm−1, F(000) = 1208. Of 17527 measured data, 9426 were unique (Rint = 0.0484) and 7615 observed (I > 2σ(I)]) to give R1 = 0.0953 and wR2 = 0.2107.
[Pd(1)Cl2], yellow prisms were obtained from a catalytic solution starting from [PdCl2(MeCN)2] and BDTBPMB (ESI Table S2, entry 2†) C24H44P2Cl2Pd, M = 571.83, triclinic, space group P21/c, a = 9.1553(15), b = 19.646(3), c = 15.159(3) Å β = 101.766(5)°, U = 2669.3(8) Å3, Z = 4, Dc = 1.423 Mgm−3, μ = 1.025 mm−1, F(000) = 1192. Of 14255 measured data, 4316were unique (Rint = 0.0241) and 3889 observed (I > 2σ(I)]) to give R1 = 0.0315 and wR2 = 0.0710, GOF = 1.091.
Conclusions
[Pd2dba3]/BDTBPMB is highly active for the methoxycarbonylation of phenylethyne (TOFo > 2000 mol product·(mol catalyst·h)−1,with very high linear selectivity towards methyl 3-phenylpropenoate (3). The by-product, dimethyl 2-phenyl-1,3-butanedioate (5) which is formed in small amounts (<10%), forms almost exclusively as a result of a second methoxycarbonylation of the branched product, methyl 2-methylpropenoate (4). This unusual selectivity has been attributed to a preference for the hydride mechanism over the carbomethoxy mechanism. The product cinnamate esters are usually made by Heck reactions. Advantageously, their synthesis from methoxycarbonylation of alkynes, some of which are available from oil refining, does not involve halides. We note, however, that alkynes are often formed by Sonogashira coupling, which usually involves halide and base, so the overall process in that case is not halide free.15The methoxycarbonylation of linear aliphatic alkynes gave predominantly α,β–unsaturated esters after short reaction times, but α,ω–diesters after longer reaction times, with high yields and good selectivity in a single cascade methoxycarbonylation–isomerisation–methoxycarbonylation sequence. The products are important building blocks for polyesters and polyamides. The internal alkyne, 4-octyne, gave only the monocarbonylated product, methyl 2-propylhex-2-enoate, because the isomerisation is blocked as a result of the high steric hindrance surrounding the double bond in this product.
Hydroxycarbonylation of phenylethyne gave mainly 3-phenyl propenoic acid when BDTBPMB was used as the ligand, but the selectivity changed towards 2-phenyl-1,4-butanedoic acid, the double carbonylated product, when the less hindered BDPPMB was employed.
Acknowledgements
We thank Lucite International (A. A. N. M) and the University of St. Andrews (L. M. R) for studentships. Special thanks are extended to Dr David Johnson for prolific discussions.Notes and references
- G. Kiss, Chem. Rev., 2001, 101, 3435 CrossRef CAS.
- (a) E. Drent, P. Arnoldy and P. H. M. Budzelaar, J. Organomet. Chem., 1994, 475, 57 CrossRef CAS; (b) E. Drent, P. Arnoldy and H. M. Budzelaar, J. Organomet. Chem., 1993, 455, 247 CrossRef CAS; (c) E. Drent, W. W. Jager and J. C. L. J. Suykerbuyk, 1995, WO95 /05357; (d) E. Drent and D. H. L. Pello, 1995, WO95 /03269; (e) E. Drent and W. W. Jager, 1998, US5719313.
- (a) A. Dervisi, P. G. Edwards, P. D. Newman, R. P. Tooze, S. J. Coles and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1999, 1113 RSC; (b) A. Dervisi, P. G. Edwards, P. D. Newman and R. P. Tooze, J. Chem. Soc., Dalton Trans., 2000, 523 RSC.
- (a) B. El Ali and H. Alper, J. Mol. Catal., 1991, 67, 29 CrossRef CAS; (b) K. Itoh, M. Miura and M. Nomura, Tetrahedron Lett., 1992, 33, 5367; (c) Y. Kushino, K. Itoh, M. Miura and M. Nomura, J. Mol. Catal., 1994, 89, 151 CrossRef CAS; (d) M. T. Reetz, R. Demuth and R. Goddard, Tetrahedron Lett., 1998, 39, 7089 CrossRef CAS; (e) A. Scrivanti, V. Beghetto, E. Campagna, M. Zanato and U. Matteoli, Organometallics, 1998, 17, 630 CrossRef CAS; (f) M. T. Reetz, R. Demuth and R. Goddard, Tetrahedron Lett., 1998, 39, 7089 CrossRef CAS; (g) A. Scrivanti, V. Beghetto, E. Campagna, M. Zanato and U. Matteoli, Organometallics, 1998, 17, 630 CrossRef CAS; (h) S. Jayasree, A. Seayad, S. P. Grupte and R. V. Chaudhari, Catal. Lett., 1999, 58, 213 CrossRef CAS; (i) A. Scrivanti, V. Beghetto, M. Zanato and U. Matteoli, J. Mol. Catal. A: Chem., 2000, 160, 331 CrossRef CAS; (j) M. Akao, S. Sugawara, K. Amino and Y. Inoue, J. Mol. Catal. A: Chem., 2000, 157, 117 CrossRef CAS; (k) A. Scrivanti, U. Matteoli, V. Beghetto, S. Antonaroli, R. Scarpelli and B. Crociani, J. Mol. Catal. A: Chem., 2001, 170, 51 CrossRef CAS; (l) M. L. Clarke, D. J. Cole-Hamilton, D. F. Foster, A. M. Z. Slawin and J. D. Woollins, J. Chem. Soc., Dalton Trans., 2002, 1618 RSC; (m) J. J. M. de Pater, E. P. Maljaars, E. de Wolf, M. Lutz, A. L. Spek, B. Deelman, C. J. Elsevier and G. Van Koten, Organometallics, 2005, 24, 5299 CrossRef CAS.
- For recent reviews, see: (a) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS; (c) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS.
- (a) W. Clegg, G. R. Eastham, M. R. J. Elsegood, R. P. Tooze, X. L. Wang and K. Whiston, Chem. Commun., 1999, 1877 RSC; (b) G. R. Eastham, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Chem. Commun., 2000, 609 RSC; (c) G. R. Eastham, R. P. Tooze, M. Kilner, D. F. Foster and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 2002, 1613 RSC; (d) W. Clegg, G. R. Eastham, M. R. J. Elsegood, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Organometallics, 2002, 21, 1832 CrossRef CAS; (e) W. Clegg, G. R. Eastham, M. R. J. Elsegood, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, J. Chem. Soc., Dalton Trans., 2002, 3300 RSC; (f) R. A. M. Robertson and D. J. Cole-Hamilton, Coord. Chem. Rev., 2002, 225, 67 CrossRef CAS; (g) D. J. Cole-Hamilton and R. A. M. Robertson in Catalytic Synthesis of Alkene-Carbon Monoxide Copolymers and Cooligomers, Ed. A. Sen, Klüwer, Dordrecht, 2003, Chapter 3, 37 Search PubMed.
- C. J. Rodriguez, D. F. Foster, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2004, 1720 RSC.
- For other examples of BDTBPMB in catalysis, see: (a) C. Jiménez, G. R. Eastham and D. J. Cole-Hamilton, Inorg. Chem. Commun., 2005, 8, 878 CrossRef CAS; (b) Y. Zhu, S. Mujcinovic, W. R. Jackson and A. J. Robinson, Green Chem., 2006, 8, 746 RSC; (c) E. Drent and R. Ernst, 2004, WO200410394 8; (d) E. Drent, R. Ernst, W. W. Jager and C. A. Krom, 2006, WO2006/084892A2; (e) J. A. M. Broekhoven, E. Drent, W. W. Jager and C. A. Krom, 2006, WO2006084889A1; (f) C. Jiménez, G. R. Eastham and D. J. Cole-Hamilton, Dalton Trans., 2005, 1826 RSC; (g) C. Jiménez-Rodriguez, P. J. Pogorzelec, G. R. Eastham, A. M. Z. Slawin and D. J. Cole-Hamilton, Dalton Trans., 2007, 4160 RSC; (h) A. A. Núñez Magro, G. R. Eastham and D. J. Cole-Hamilton, Dalton Trans., 2009, 4683 RSC; (i) G. R. Eastham, M. Waugh and P. I. Richards, 2008, WO2008075108.
- (a) A. J. Rucklidge, G. E. Morris and D. J. Cole-Hamilton, Chem. Commun., 2005, 1176 RSC; (b) H. Ooka, T. Inoue, S. Itsumo and M. Tanaka, Chem. Commun., 2005, 1173 RSC; (c) A. J. Rucklidge, G. E. Morris, A. M. Z. Slawin and D. J. Cole-Hamilton, Helv. Chim. Acta, 2006, 89, 1783 CrossRef CAS.
- For another example of the use of an air-stable phosphonium salt, which has been used to synthesise phosphine complexes, see: M. R. Netherton and G. C. Fu, Org. Lett., 2001, 26, 4295 Search PubMed.
- A. A. Nùñez-Magro, PhD Thesis, University of St. Andrews, 2007.
- C. J. Mathews, P. J. Smith and T. Welton, J. Mol. Catal. A: Chem., 2003, 206, 77 CrossRef CAS.
- D. J. Morris, G. Docherty, G. Woodward and M. Wills, Tetrahedron Lett., 2007, 48, 949 CrossRef CAS.
- M. L. Clarke, Tetrahedron Lett., 2004, 45, 4043 CrossRef CAS.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of reactions involving alternative ligands, metal precursors and alcohols as well as full crystallographic details for 1. [BDTBPMBH2]BF4 and [Pd(1)Cl2]. CCDC reference numbers 774479–774481. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00276c |
| This journal is © The Royal Society of Chemistry 2010 |